Orthopedic Design & Technology Magazine




March/April 2024
-
Extreme Fixes for Orthopedic Extremity Surgery
Digital solutions like AI, patient-specific smart implants, and surgical robotics are creating new opportunities in the rapidly evolving extremities market.03.19.24
-

Print Shop: Insights on Orthopedic Additive Manufacturing
Investigating the achievements, benefits, and limitations of additive manufacturing for orthopedic implants and instruments.03.19.24
-

Molding Is Taking Its Place in Orthopedic Device Manufacturing
As molders continue to expand capabilities attractive to orthopedic device makers, the fabrication process is growing in use.03.19.24
-
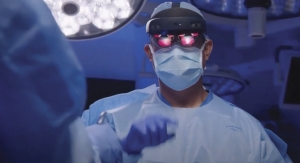
A View of Mixed-Reality Tech's Future in Orthopedics
With mixed-reality headsets, orthopedic surgeons can improve procedural precision, streamline workflow, and democratize care.03.19.24
-

Improving Patient-Specific Outcomes with In-Silico Clinical Trials
In-silico testing helps evaluate implant designs for personalized treatment.03.19.24
-
 Regulatory PerspectivesCMC Considerations for Medical Device-Led Combination ProductsCreating antibiotic-loaded orthopedic combination products demands meticulous yet distinct CMC considerations.03.19.24
Regulatory PerspectivesCMC Considerations for Medical Device-Led Combination ProductsCreating antibiotic-loaded orthopedic combination products demands meticulous yet distinct CMC considerations.03.19.24
-
 Orthopedic InsightsIndustry Dynamics from AAOS 2024A closer examination of some key orthopedic device industry battlegrounds.03.19.24
Orthopedic InsightsIndustry Dynamics from AAOS 2024A closer examination of some key orthopedic device industry battlegrounds.03.19.24
-
 Market SnapshotU.S. Obesity Versus Hip and Knee ReplacementsThe connection between the increase in weight is correlated to an increase in annual medical costs.03.19.24
Market SnapshotU.S. Obesity Versus Hip and Knee ReplacementsThe connection between the increase in weight is correlated to an increase in annual medical costs.03.19.24
-
 The Last WordOnward and Upward in Treating Spinal Cord InjuryOnward Medical's ARC therapy delivers targeted, programmed spinal cord stimulation to restore movement and other functions in people with spinal cord injury.03.19.24
The Last WordOnward and Upward in Treating Spinal Cord InjuryOnward Medical's ARC therapy delivers targeted, programmed spinal cord stimulation to restore movement and other functions in people with spinal cord injury.03.19.24
-
 EditorialCelebrating Change in the Orthopedic Device IndustryMost sessions at this year’s ODT Forum will discuss the changes taking place within the industry.03.19.24
EditorialCelebrating Change in the Orthopedic Device IndustryMost sessions at this year’s ODT Forum will discuss the changes taking place within the industry.03.19.24
-
 ColumnsHow Medical Manufacturers Can Practice Due Diligence for Change ControlA broader view of design and process changes and the implications of those changes is critical to both the product’s and company’s continued success.03.19.24
ColumnsHow Medical Manufacturers Can Practice Due Diligence for Change ControlA broader view of design and process changes and the implications of those changes is critical to both the product’s and company’s continued success.03.19.24
-
 ColumnsBoost Mobility: Precision Joint Replacement with 3D-Printed Implants3D printing enables manufacturers and surgeons to approach arthroplasty in a whole new way.03.19.24
ColumnsBoost Mobility: Precision Joint Replacement with 3D-Printed Implants3D printing enables manufacturers and surgeons to approach arthroplasty in a whole new way.03.19.24
