12.01.08
Preparing for FDA BIMO Inspections
An industry expert shares tips and useful data for developing a quality-systems approach for conducting medical device clinical trials.
Susan M. Rockwell
Over the past five years, the FDA's Division of Bioresearch Monitoring (BIMO), in the Office of Compliance at the Center for Devices and Radiological Health (CDRH), has conducted, on average, 325 to 350 inspections of medical device clinical trials each year. These inspections mainly are divided among the clinical investigators, the Institutional Review Boards (IRBs), and the medical device sponsors.
Typically, half of all BIMO inspections occur at the clinical investigational site, with the rest split between the medical device sponsors and the IRBs. A small number of inspections also occur each year at clinical laboratories.
The most important step in preparing for an FDA BIMO inspection is to develop a quality systems approach for conducting medical device clinical trials. |
Because FDA's BIMO inspections can occur at any time during the clinical trial, it is imperative for medical device sponsors to be prepared from the very beginning. The first—and most important—step in preparing for an FDA BIMO inspection is to develop a quality-systems approach for conducting medical device clinical trials.
The agency's BIMO division has outlined four ongoing global issues for the medical device industry to focus on (based on CDRH Compliance Actions and Inspectional Data, 2006):
- Systems to identify and/or address regulatory shortcomings
- Systems to correct and/or prevent recurring issues
- Accountable company culture
- Environment of conflict of interest
As part of a company's quality plan, there should be systems in place to correct or prevent recurring issues.
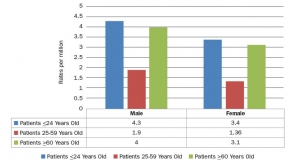
Listed in Table 1 (above) are some of the most prevalent reported sponsor deficiencies over an eight-year period of FDA BIMO inspections.
In addition to the four deficiencies listed in Table 1, BIMO inspectors also reported the following sponsor deficiencies in 2007:
- Failure to submit Progress Reports (36%)
- Inadequate unanticipated adverse device effects analysis & reporting (27%)
- Failure to inform investigators (21%)
- Failure to obtain signed Investigator Agreement (15%)
- Unqualified monitors (12%)
In addition to the five deficiences shown in Table 1 and Table 2, there needs to be more ownership by company officials or stakeholders in positions of authority—either within a medical device company or at a clinical investigational site. Although noncompliance rates had been improving steadily for sponsors during the past few years, noncompliance rates jumped significantly in 2007. Although the increase in noncompliance rates could be an anomaly, the FDA believes it may be due to an increase in For Cause Inspections at sponsor companies. For Cause Inspections, conducted as a follow-up to a complaint, typically find serious deficiencies or departures from the regulations. Ignoring serious deficiencies, or not taking ownership of these problems, can result in a laundry list of regulatory and administrative actions, including:
- Warning letter
- Re-inspection
- Rejection of study data
- Investigational device exemption termination or withdrawal of premarket approval application
- Disqualification of clinical investigators or IRBs
- Civil money penalties
- Prosecution
- Medical device clinical trial sponsors need to promote a quality-system approach to the conduct of their clinical trials:
- Creating SOPs for clinical study functions
- Hiring employees with experience in conducting clinical studies
- Training employees, contractors, and clinical investigators appropriately and holding them accountable
- Conducting internal audits of clinical processes and procedures
- Increasing senior management oversight of clinical study progress