02.11.13
In December, the Internal Revenue Service (IRS) published its final guidance on the medical device excise tax that took effect Jan. 1 under the Patient Protection and Affordable Care Act. The IRS composed the guidance after consulting with technical experts at the U.S. Food and Drug Administration (FDA) as well as reviewing public comment on the issues.
The document comprises 58 pages explaining such things as the definition of a taxable medical device and tax payment options for companies.
Generally, under the final regulations, a taxable medical device is one that is listed as a device with the FDA. If a device is not listed with the agency, but the FDA determines it should have been, that device will be treated as such, effective of the date the FDA notifies the manufacturer or importer that corrective action with respect to its listing is required.
Conversely, if a manufacturer lists a device with the FDA but the device was not required to be listed, a credit or refund may be available for tax paid on sales of the device once the device has been de-listed.
The guidance includes a “retail exemption,” which exempts devices typically purchased by the general public at retail for individual use such as eyeglasses, contact lenses, and hearing aids. There also are some devices listed with the FDA that qualify for the IRS’ retail exemption “safe harbor.” These include devices in the agency’s in-vitro diagnostics home use lab test database, devices the FDA considers over-the-counter. Some prosthetics and orthotics are included in this exemption as well.
According to the IRS, sales of taxable medical devices for further manufacture or export may be made tax free if certain registration and other requirements are met.
Perhaps making the tax more of an inevitable reality than ever, device companies now have a solid payment schedule set forth by the tax agency. The first quarterly return for the device excise tax is due April 30, 2013, for the months of January, February and March, and can be filed on paper or electronically. Manufacturers and importers of taxable medical devices generally are required to make semi-monthly deposits of tax. The first semi-monthly deposit for the medical device excise tax, which covers the first 15 days of January, was due Jan. 29. In general, manufacturers and importers must use electronic funds transfer to make excise tax deposits through the electronic federal tax payment system. Medtech advocacy organization the Advanced Medical Technology Association (AdvaMed) still insists the device excise tax should be repealed. “AdvaMed is carefully reviewing the regulations released by the IRS,” President and CEO Stephen Ubl said in a prepared statement. “But regardless, Congress should act to address this $30 billion tax. There is strong bipartisan support for action... This tax could push us off an innovation cliff, costing as many as 43,000 jobs and hurting the ability of medical technology companies to find tomorrow’s treatments and cures. It should be repealed.”
Ubl went on to mention the layoffs and research cutbacks companies have been making, ostensibly in anticipation of the tax. “The tax also is scaring away investors and it threatens U.S. leadership in medical technology, where America is a net exporter to the tune of $5.4 billion a year,” he added.
“This final rule does nothing to prevent the loss of jobs and innovation that has already occurred as a result of the medical device tax, and will unfortunately continue if we do not repeal this bad policy,” agreed Mark Leahey, president and CEO of the Medical Device Manufacturers Association.
Medtech Industry Unhappy About Tax Repeal Defeat
2013 started off with a bang, but it wasn’t the sound of the United States falling off the fiscal cliff. The U.S. Congress managed to make a deal regarding the expiration of tax cuts enacted under the last Bush administration, but failed to repeal or delay the medical device excise tax.
“The effort to repeal the medical device tax will continue,” vowed Stephen Ubl, president, CEO of the Advanced Medical Technology Association (Advamed). “The passage of a scaled-back fiscal cliff package that did not address the medical device tax does not diminish the need to repeal the tax. It also does not diminish the bipartisan support for the repeal effort, which is premised on the recognition that the tax is costing jobs and threatening patient care. We urge Congress to repeal the device tax as it returns to address the other pressing tax and budget issues facing the country, so that we can avoid going over the medical technology innovation cliff.”
The “fiscal cliff package” includes items such as the stoppage of a 27 percent reduction in payments to Medicare providers, which is beneficial to Medicare patients who were at risk of being dropped by care providers unable to afford the cuts in compensation. However, the package did cut compensation for some specific services. The package reduces Medicare payments to physicians’ offices for advanced imaging services by $800 million and hospital reimbursements for radiation therapy by $300 million over 10 years. This, on top of the more than $1 billion in cuts for imaging and radiation therapy services put in place earlier in 2012, places what Ubl sees as unacceptable strain on the social insurance program.
“AdvaMed also is disappointed in the cuts to imaging and diabetes-related services that are a part of the package passed by Congress,” Ubl said. “We believe these cuts will hurt access to important technologies that Medicare beneficiaries depend upon.”
The Medical Imaging & Technology Alliance (MITA) added its voice to the chorus, criticizing the cuts to Medicare imaging compensation. “The House should reject Medicare cuts since the data clearly show that imaging use per beneficiary is on the decline,” said Gail Rodriguez, MITA’s executive director. “It is arbitrary and capricious for Congress to cut imaging and radiation therapy reimbursements without a full understanding of how those cuts negatively impact their constituents’ ability to receive imaging and radiation therapy that saves lives.”
Rodriguez also drew focus to lawmakers’ failure to delay the device tax. “When you add up all the Medicare cuts and Congress’ reluctance to address the $30 billion medical device tax, this legislation produces a devastating impact that harms patient access to care, moves manufacturing jobs overseas and threatens America’s leadership in medical research and development. We hope the Administration and congressional leaders will take notice of the growing bipartisan opposition to the job-killing device tax and immediately repeal or delay it.”
Stronger Regulation for MoM Hip Implants Proposed by FDA
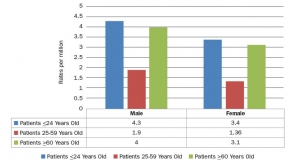
Metal-on-metal (MoM) hip implants are more or less accepted as a risky choice due to their tendency to deposit miniscule metal fragments into the bloodstream. According to an explanation from the U.S. Food and Drug Administration (FDA), over time, the metal particles around some MoM implants can cause damage to bone and/or tissue surrounding the implant and joint. This often is called an adverse local tissue reaction or an adverse reaction to metal debris.
Because MoM implants were marketed in the United States before the 1976 legislation that gave the FDA premarket authority over medical devices, they were classified as “preamendment devices.” Therefore, they were designated Class III (higher risk) devices but remained regulated under the 510(k) premarket notification program. On Jan. 17, the FDA issued a proposed order requiring manufacturers of MoM total hip replacement systems to submit premarket approval applications as opposed to 510(k) clearance applications.
The FDA also released an updated report on MoM hip implants that claims the implants can cause soft-tissue damage and pain, possibly prompting additional surgery. The report is based on reviews of new published literature and the results of a June 2012 Orthopedic and Rehabilitation Devices Advisory Panel meeting. According to the report, soft tissue damage may lead to pain, implant loosening, device failure and the need for revision surgery (a surgical procedure where the implant is replaced). Some of the metal ions (commonly cobalt or chromium) released enter the bloodstream and travel to other parts of the body, where they may cause symptoms or illnesses. The FDA, however, added a caveat—there is not sufficient scientific data to specify exactly how high the level of ions need to be to trigger adverse systemic effects. Current and recent patients have reacted differently to metal wear particles.
FDA recommends that orthopedic surgeons seriously weigh the benefit-risk profile of using a MoM implant rather than an alternative such as metal-on-polyethylene, ceramic-on-polyethylene, ceramic-on-ceramic or ceramic-on-metal. Surgeons are encouraged to inform patients of all risks associated with MoM implants, and to monitor patient populations for MoM implants that are contraindicated.
MoM implant patients with systemic symptoms are more likely to visit their primary care practitioner than their orthopedic surgeon, so the FDA stresses the importance that all health care providers be aware of metal ion adverse events that may occur in MoM hip implant patients. Based on case reports, these events may include:
The agency launched a website in February 2011 to keep physicians and patients abreast of new information regarding MoM implants and informed about the general risks associated with them.
FDA to Hold Workshop on Medical Device Labeling
The U.S. Food and Drug Administration (FDA) has announced a public workshop to be held on April 29-30. Under discussion will be standardizing the format and content of medical device labeling and the development of a public repository or database that would provide access to medical device labeling. Regulators said the agency is concerned that the lack of standardized content and format and access to medical device labeling may increase the risk of medical errors. But by the same token, standardization and public access to medical device labeling raises several potential regulatory concerns (e.g., the ability to make modifications to the labeling) and could have product liability implications. The FDA recommends that clients pay close attention to developments on the issue and consider submitting comments.
Device “labeling” refers to any printed material on any medical device or any of its containers or wrappers, or on any material that accompanies a device such as a guide or manual.
The meeting, held at the FDA’s White Oak campus in Silver Spring, Md., will be webcast. The agency is seeking input and comments on the following topics:
The document comprises 58 pages explaining such things as the definition of a taxable medical device and tax payment options for companies.
Generally, under the final regulations, a taxable medical device is one that is listed as a device with the FDA. If a device is not listed with the agency, but the FDA determines it should have been, that device will be treated as such, effective of the date the FDA notifies the manufacturer or importer that corrective action with respect to its listing is required.
Conversely, if a manufacturer lists a device with the FDA but the device was not required to be listed, a credit or refund may be available for tax paid on sales of the device once the device has been de-listed.
The guidance includes a “retail exemption,” which exempts devices typically purchased by the general public at retail for individual use such as eyeglasses, contact lenses, and hearing aids. There also are some devices listed with the FDA that qualify for the IRS’ retail exemption “safe harbor.” These include devices in the agency’s in-vitro diagnostics home use lab test database, devices the FDA considers over-the-counter. Some prosthetics and orthotics are included in this exemption as well.
According to the IRS, sales of taxable medical devices for further manufacture or export may be made tax free if certain registration and other requirements are met.
Perhaps making the tax more of an inevitable reality than ever, device companies now have a solid payment schedule set forth by the tax agency. The first quarterly return for the device excise tax is due April 30, 2013, for the months of January, February and March, and can be filed on paper or electronically. Manufacturers and importers of taxable medical devices generally are required to make semi-monthly deposits of tax. The first semi-monthly deposit for the medical device excise tax, which covers the first 15 days of January, was due Jan. 29. In general, manufacturers and importers must use electronic funds transfer to make excise tax deposits through the electronic federal tax payment system. Medtech advocacy organization the Advanced Medical Technology Association (AdvaMed) still insists the device excise tax should be repealed. “AdvaMed is carefully reviewing the regulations released by the IRS,” President and CEO Stephen Ubl said in a prepared statement. “But regardless, Congress should act to address this $30 billion tax. There is strong bipartisan support for action... This tax could push us off an innovation cliff, costing as many as 43,000 jobs and hurting the ability of medical technology companies to find tomorrow’s treatments and cures. It should be repealed.”
Ubl went on to mention the layoffs and research cutbacks companies have been making, ostensibly in anticipation of the tax. “The tax also is scaring away investors and it threatens U.S. leadership in medical technology, where America is a net exporter to the tune of $5.4 billion a year,” he added.
“This final rule does nothing to prevent the loss of jobs and innovation that has already occurred as a result of the medical device tax, and will unfortunately continue if we do not repeal this bad policy,” agreed Mark Leahey, president and CEO of the Medical Device Manufacturers Association.
Medtech Industry Unhappy About Tax Repeal Defeat
2013 started off with a bang, but it wasn’t the sound of the United States falling off the fiscal cliff. The U.S. Congress managed to make a deal regarding the expiration of tax cuts enacted under the last Bush administration, but failed to repeal or delay the medical device excise tax.
“The effort to repeal the medical device tax will continue,” vowed Stephen Ubl, president, CEO of the Advanced Medical Technology Association (Advamed). “The passage of a scaled-back fiscal cliff package that did not address the medical device tax does not diminish the need to repeal the tax. It also does not diminish the bipartisan support for the repeal effort, which is premised on the recognition that the tax is costing jobs and threatening patient care. We urge Congress to repeal the device tax as it returns to address the other pressing tax and budget issues facing the country, so that we can avoid going over the medical technology innovation cliff.”
The “fiscal cliff package” includes items such as the stoppage of a 27 percent reduction in payments to Medicare providers, which is beneficial to Medicare patients who were at risk of being dropped by care providers unable to afford the cuts in compensation. However, the package did cut compensation for some specific services. The package reduces Medicare payments to physicians’ offices for advanced imaging services by $800 million and hospital reimbursements for radiation therapy by $300 million over 10 years. This, on top of the more than $1 billion in cuts for imaging and radiation therapy services put in place earlier in 2012, places what Ubl sees as unacceptable strain on the social insurance program.
“AdvaMed also is disappointed in the cuts to imaging and diabetes-related services that are a part of the package passed by Congress,” Ubl said. “We believe these cuts will hurt access to important technologies that Medicare beneficiaries depend upon.”
The Medical Imaging & Technology Alliance (MITA) added its voice to the chorus, criticizing the cuts to Medicare imaging compensation. “The House should reject Medicare cuts since the data clearly show that imaging use per beneficiary is on the decline,” said Gail Rodriguez, MITA’s executive director. “It is arbitrary and capricious for Congress to cut imaging and radiation therapy reimbursements without a full understanding of how those cuts negatively impact their constituents’ ability to receive imaging and radiation therapy that saves lives.”
Rodriguez also drew focus to lawmakers’ failure to delay the device tax. “When you add up all the Medicare cuts and Congress’ reluctance to address the $30 billion medical device tax, this legislation produces a devastating impact that harms patient access to care, moves manufacturing jobs overseas and threatens America’s leadership in medical research and development. We hope the Administration and congressional leaders will take notice of the growing bipartisan opposition to the job-killing device tax and immediately repeal or delay it.”
Stronger Regulation for MoM Hip Implants Proposed by FDA
Metal-on-metal (MoM) hip implants are more or less accepted as a risky choice due to their tendency to deposit miniscule metal fragments into the bloodstream. According to an explanation from the U.S. Food and Drug Administration (FDA), over time, the metal particles around some MoM implants can cause damage to bone and/or tissue surrounding the implant and joint. This often is called an adverse local tissue reaction or an adverse reaction to metal debris.
Because MoM implants were marketed in the United States before the 1976 legislation that gave the FDA premarket authority over medical devices, they were classified as “preamendment devices.” Therefore, they were designated Class III (higher risk) devices but remained regulated under the 510(k) premarket notification program. On Jan. 17, the FDA issued a proposed order requiring manufacturers of MoM total hip replacement systems to submit premarket approval applications as opposed to 510(k) clearance applications.
The FDA also released an updated report on MoM hip implants that claims the implants can cause soft-tissue damage and pain, possibly prompting additional surgery. The report is based on reviews of new published literature and the results of a June 2012 Orthopedic and Rehabilitation Devices Advisory Panel meeting. According to the report, soft tissue damage may lead to pain, implant loosening, device failure and the need for revision surgery (a surgical procedure where the implant is replaced). Some of the metal ions (commonly cobalt or chromium) released enter the bloodstream and travel to other parts of the body, where they may cause symptoms or illnesses. The FDA, however, added a caveat—there is not sufficient scientific data to specify exactly how high the level of ions need to be to trigger adverse systemic effects. Current and recent patients have reacted differently to metal wear particles.
FDA recommends that orthopedic surgeons seriously weigh the benefit-risk profile of using a MoM implant rather than an alternative such as metal-on-polyethylene, ceramic-on-polyethylene, ceramic-on-ceramic or ceramic-on-metal. Surgeons are encouraged to inform patients of all risks associated with MoM implants, and to monitor patient populations for MoM implants that are contraindicated.
MoM implant patients with systemic symptoms are more likely to visit their primary care practitioner than their orthopedic surgeon, so the FDA stresses the importance that all health care providers be aware of metal ion adverse events that may occur in MoM hip implant patients. Based on case reports, these events may include:
- General hypersensitivity reaction (skin rash);
- Cardiomyopathy;
- Neurological changes including sensory changes (auditory or visual impairments, for example);
- Psychological status change (including depression);
- Renal function impairment; and
- Thyroid dysfunction.
The agency launched a website in February 2011 to keep physicians and patients abreast of new information regarding MoM implants and informed about the general risks associated with them.
FDA to Hold Workshop on Medical Device Labeling
The U.S. Food and Drug Administration (FDA) has announced a public workshop to be held on April 29-30. Under discussion will be standardizing the format and content of medical device labeling and the development of a public repository or database that would provide access to medical device labeling. Regulators said the agency is concerned that the lack of standardized content and format and access to medical device labeling may increase the risk of medical errors. But by the same token, standardization and public access to medical device labeling raises several potential regulatory concerns (e.g., the ability to make modifications to the labeling) and could have product liability implications. The FDA recommends that clients pay close attention to developments on the issue and consider submitting comments.
Device “labeling” refers to any printed material on any medical device or any of its containers or wrappers, or on any material that accompanies a device such as a guide or manual.
The meeting, held at the FDA’s White Oak campus in Silver Spring, Md., will be webcast. The agency is seeking input and comments on the following topics:
- Summary of the FDA’s work on medical device labeling;
- Standard content and format of device labeling; and
- Repository of medical device labeling for home use devices.