Sam Brusco, Associate Editor03.16.21
The FDA has advised about higher than expected risk of the polyethylene (plastic) component of the Scandinavian Total Ankle Replacement (STAR Ankle) breaking as early as three to four years after implantation. This may lead to surgery to repair or replace the device.
FDA’s recent analysis of FDA-required post approval studies and adverse event reports have shown the potential risk of the component breaking for all STAR Ankle devices, regardless of the manufacture or distribution date.
FDA believes the STAR Ankle remains appropriate for older patients with lower activity levels. Based on additional medical literature and real-world evidence, patients with more active lifestyles, osteoarthritis, or age below 55 years old may have higher risk of the plastic component breaking.
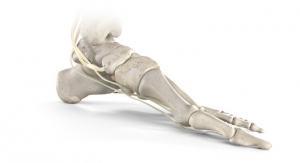
The STAR Ankle is indicated for use as a non-cemented total ankle prosthetic and used to replace a painful arthritic ankle joint due to osteoarthritis, post-traumatic arthritis, or rheumatoid arthritis. The STAR Ankle is comprised of a tibial plate, a mobile-bearing polyethylene component, and a talar component. It’s designed to allow for some normal ankle mobility and function. The mobile-bearing device component is made from sterilized polyethylene.
This device was approved in the Premarket Approval (PMA) application, P050050 with two post-approval studies:
Based on the long-term post-approval study results, the plastic component fractured at a cumulative rate of 13.8 percent (12/87) eight-years after implantation. All fractures required additional surgery. Fractures were also observed as early as three to four years after implantation. The higher fracture rate and earlier than expected occurrence are concerning when compared with other comparable total ankle replacement devices, and may also be underestimated as two polyethylene fractures were not diagnosed until surgical exploration in this study.
Stryker has previously acknowledged and communicated concern about fracture rate in the post-approval study. However, Stryker’s communication excluded devices manufactured after August 1, 2014, when changes were made in the inner-pouch foil packaging to limit material degradation of the polyethylene component.
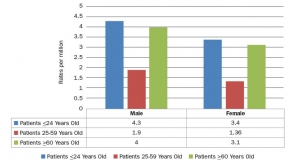
Since 2009, at least 1,841 adverse event reports have been received for the STAR Ankle. The FDA concluded about 300 of these reports described events of fractured plastic components and included STAR Ankle devices manufactured before and after the 2014 packaging change.
Plastic STAR Ankle component fractures may be attributed to multiple factors, including component thickness, material degradation, surgical factors, and patient factors—such as younger patients with higher activity levels. The long-term fracture rate is not known in devices manufactured after the 2014 packaging change.
FDA’s recent analysis of FDA-required post approval studies and adverse event reports have shown the potential risk of the component breaking for all STAR Ankle devices, regardless of the manufacture or distribution date.
FDA believes the STAR Ankle remains appropriate for older patients with lower activity levels. Based on additional medical literature and real-world evidence, patients with more active lifestyles, osteoarthritis, or age below 55 years old may have higher risk of the plastic component breaking.
The STAR Ankle is indicated for use as a non-cemented total ankle prosthetic and used to replace a painful arthritic ankle joint due to osteoarthritis, post-traumatic arthritis, or rheumatoid arthritis. The STAR Ankle is comprised of a tibial plate, a mobile-bearing polyethylene component, and a talar component. It’s designed to allow for some normal ankle mobility and function. The mobile-bearing device component is made from sterilized polyethylene.
This device was approved in the Premarket Approval (PMA) application, P050050 with two post-approval studies:
- The first post-approval study—an eight-year follow-up of a cohort of patients who were enrolled before the device was approved in 2009.
- The second post-approval study—a new enrollment, prospective, multicenter, single arm two-year study of the same device to examine performance compared to the first study. The second study included patients with and without the new packaging. Of note, this communication does not include the results from this post-approval study due to its short follow-up.
Based on the long-term post-approval study results, the plastic component fractured at a cumulative rate of 13.8 percent (12/87) eight-years after implantation. All fractures required additional surgery. Fractures were also observed as early as three to four years after implantation. The higher fracture rate and earlier than expected occurrence are concerning when compared with other comparable total ankle replacement devices, and may also be underestimated as two polyethylene fractures were not diagnosed until surgical exploration in this study.
Stryker has previously acknowledged and communicated concern about fracture rate in the post-approval study. However, Stryker’s communication excluded devices manufactured after August 1, 2014, when changes were made in the inner-pouch foil packaging to limit material degradation of the polyethylene component.
Since 2009, at least 1,841 adverse event reports have been received for the STAR Ankle. The FDA concluded about 300 of these reports described events of fractured plastic components and included STAR Ankle devices manufactured before and after the 2014 packaging change.
Plastic STAR Ankle component fractures may be attributed to multiple factors, including component thickness, material degradation, surgical factors, and patient factors—such as younger patients with higher activity levels. The long-term fracture rate is not known in devices manufactured after the 2014 packaging change.