James A. Dunning, Owner, QPC Services LLC05.19.17
After years of working in and consulting for pharmaceutical and medical device companies on quality and regulatory matters, I am still somewhat surprised by the difficulty both industries have in understanding the other’s Current Good Manufacturing Practices (CGMP) requirements. In some cases, the respective CGMP requirements are perceived to be inferior to those with which companies are most familiar. I tend to think of the two industries’ CGMP requirements as two different, but similar languages.
There are some interesting questions regarding pharmaceutical and medical device CGMP requirements. Here’s one: Could a pharmaceutical CGMP expert evaluate a medical device company’s CGMP compliance status using Part 820 as a checklist? Likewise, could a medical device CGMP expert evaluate a pharmaceutical firm’s CGMP compliance using Part 820? There are practical reasons why this particular strategy would fail—mainly because any citation for finding would be incorrect. The match between the CGMP expert and the company’s products is key to a successful outcome.
Another concern involves the differences between the CGMP requirements for pharmaceuticals and medical devices. Is one better than the other? Is one more stringent or more technical than the other? I’m not going to answer these questions; rather, I encourage readers to ponder them, at least for a minute or two, and draw their own conclusions.
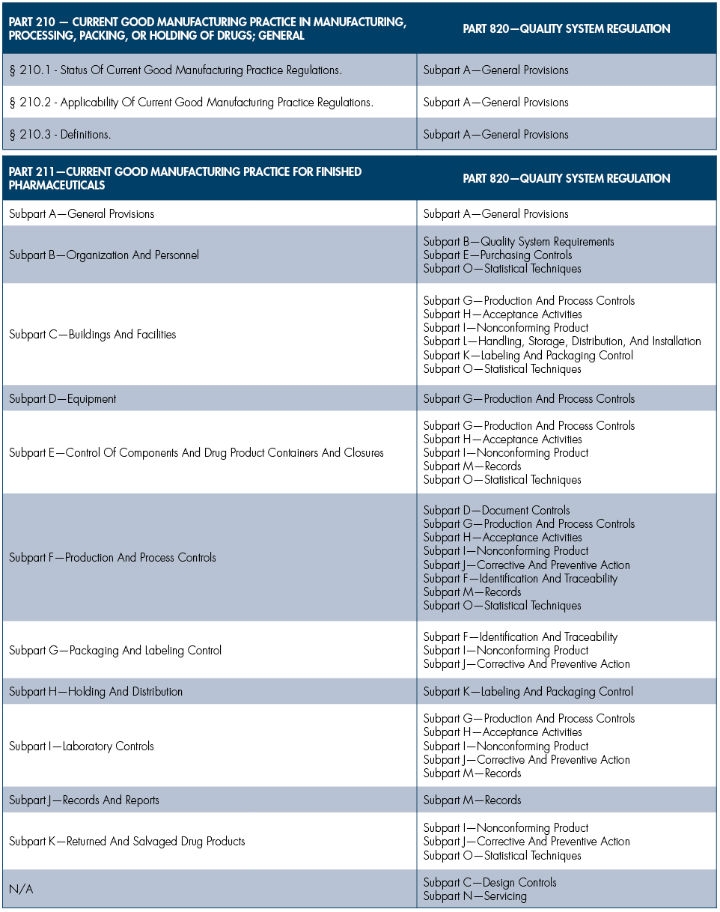
In the next several columns, I will compare sections of the pharmaceutical industry’s CGMP, Parts 210 and 211, with the medical device sector’s CGMP Part 820. The medical device CGMPs will be compared to the pharmaceutical CGMPs, with the assessment being one-directional for simplicity. The following restrictions will apply:
To easily illustrate the similarities and differences between the two industries’ CGMP requirements, I will use a basic side-by-side comparison chart with commentary. The commentary should be minimal, since showing the comparable subsections should be self-explanatory.
To get started, I will focus on the high-level comparison. In subsequent columns, I will dive deeper into the regulatory requirements.
Obviously, the overview is quite simple and easy to understand. Future columns that focus on two or three subparts per column will provide greater detail on the differences and similarities between CGMP requirements between the pharmaceutical and medtech industries. Stay tuned.
James A. “Jim” Dunning’s consulting career began in 2001. He has provided quality and regulatory consulting services for various companies ranging from Fortune 500 medical device firms to startups. Dunning’s passion, however, lies with startups and small companies, especially those in regulatory distress. He has amassed significant experience in preparing 510(k) applications, developing complete Quality Management Systems, providing Quality System Training, and advising on quality, business, and leadership issues. Dunning is a senior member of the American Society for Quality (ASQ) and a member of the Regulatory Affairs Professional Society (RAPS), the largest global organization of and for those involved with the regulation of healthcare and related products, including medical devices, pharmaceuticals, biologics, and nutritional products. He can be reached at jdunning@qpcservices.com.
There are some interesting questions regarding pharmaceutical and medical device CGMP requirements. Here’s one: Could a pharmaceutical CGMP expert evaluate a medical device company’s CGMP compliance status using Part 820 as a checklist? Likewise, could a medical device CGMP expert evaluate a pharmaceutical firm’s CGMP compliance using Part 820? There are practical reasons why this particular strategy would fail—mainly because any citation for finding would be incorrect. The match between the CGMP expert and the company’s products is key to a successful outcome.
Another concern involves the differences between the CGMP requirements for pharmaceuticals and medical devices. Is one better than the other? Is one more stringent or more technical than the other? I’m not going to answer these questions; rather, I encourage readers to ponder them, at least for a minute or two, and draw their own conclusions.
In the next several columns, I will compare sections of the pharmaceutical industry’s CGMP, Parts 210 and 211, with the medical device sector’s CGMP Part 820. The medical device CGMPs will be compared to the pharmaceutical CGMPs, with the assessment being one-directional for simplicity. The following restrictions will apply:
- Supporting standards such as risk management standards (ISO 1491 and ISO 31000) will not be considered. Nor will standards like ISO 13485 quality systems for medical devices.
- Guidances/guidelines will also not be considered.
- Part 820.30—design controls for medical devices will not be considered, since pharmaceutical research and development/product development is not addressed in the pharmaceutical CGMP regulations.
- Regulatory applications will not be considered.
- International regulatory requirements will not be considered.
- “Industry standards” will not be considered.
To easily illustrate the similarities and differences between the two industries’ CGMP requirements, I will use a basic side-by-side comparison chart with commentary. The commentary should be minimal, since showing the comparable subsections should be self-explanatory.
To get started, I will focus on the high-level comparison. In subsequent columns, I will dive deeper into the regulatory requirements.
Obviously, the overview is quite simple and easy to understand. Future columns that focus on two or three subparts per column will provide greater detail on the differences and similarities between CGMP requirements between the pharmaceutical and medtech industries. Stay tuned.
James A. “Jim” Dunning’s consulting career began in 2001. He has provided quality and regulatory consulting services for various companies ranging from Fortune 500 medical device firms to startups. Dunning’s passion, however, lies with startups and small companies, especially those in regulatory distress. He has amassed significant experience in preparing 510(k) applications, developing complete Quality Management Systems, providing Quality System Training, and advising on quality, business, and leadership issues. Dunning is a senior member of the American Society for Quality (ASQ) and a member of the Regulatory Affairs Professional Society (RAPS), the largest global organization of and for those involved with the regulation of healthcare and related products, including medical devices, pharmaceuticals, biologics, and nutritional products. He can be reached at jdunning@qpcservices.com.