Michael Barbella, Managing Editor11.21.17
Call it the Year of Resolution.
The medtech industry showed steely resolve this year in the wake of political chaos and meteorological mayhem, shortening supply chains and streamlining operations to weather the impacts of a tyrannical hurricane trifecta, and maintaining the fight for full repeal of the 2.3 percent medical device tax.
That battle proved challenging, as the controversial levy became lost among a litany of woes endangering President Donald Trump’s legislative agenda, among them: multiple investigations into Russian interference in the 2016 election; former FBI Director James Comey’s controversial firing; staff infighting; the ban on transgender military personnel; high-profile resignations; the president’s word war with North Korean leader Kim Jong-un; and vacillating tensions with Congress.
Despite all the turmoil, the industry never strayed from its goal, though it did eventually adjust its expectations. “There are opportunities—fewer and fewer though to get full repeal,” Clayton Hall, vice president of government affairs for the Medical Device Manufacturers Association, told a Medmarc Insurance Group webinar audience in August. “The likeliest outcome right now is probably an additional suspension. But there are some opportunities to get full repeal. And that’s what we’re fighting for.”
Medtech’s mettle also manifested itself in the sector’s sustained progression toward value-based care, improved payer engagement, data analytics, and digital health. The U.S. Food and Drug Administration (FDA) helped champion the latter cause in September with a first-of-its kind program designed to revolutionize digital health regulation in the United States. The FDA’s Pre-cert pilot program is intended to develop an approach toward digital health technology by looking at the software developer or digital health technology developer, rather than primarily at the product. Participants in the FDA’s precertification pilot program (FDA Pre-cert) include Apple, Fitbit, Johnson & Johnson, Pear Therapeutics, Phosphorus, Roche, Samsung, Tidepool, and Verily.
“Our method for regulating digital health products must recognize the unique and iterative characteristics of these products,” FDA Commissioner Scott Gottlieb said in launching the program. “We need to modernize our regulatory framework so that it matches the kind of innovation we’re being asked to evaluate, and helps foster beneficial technology while ensuring that consumers have access to high-quality, safe and effective digital health devices.”
Besides propelling its assimilation of digital health in 2017, the device industry’s resolve also brought resolution to a number of longstanding issues. Zimmer Biomet Holdings Inc. and Orthofix International N.V., for example, put various federal investigations behind them, while Exactech ended its 32-year run as a public company. And just in the nick of time, the president signed a new medical device user fee agreement into law, capping two years of often tense negotiations between the industry and FDA.
Insight into these and other defining moments of 2017 can be gleaned from Orthopedic Design & Technology’s annual trip down memory lane. Enjoy the journey.
Less Volume, More Value
Spectrum Health is finding it hard to let go of tradition.
Despite all its innovations in telehealth and digital medicine, the non-profit healthcare system in Western Michigan still manages its supply chain the old-fashioned way—with paper and pencil.
Not surprisingly, such an archaic method has spawned more than its fair share of old-fashioned headaches, namely, keeping abreast of product supply levels and value. Earlier this year, however, the 12-hospital network began modernizing its supply chain management program through a Johnson & Johnson initiative designed to help health systems navigate value-based care.
Using data analytics to identify “critical interdependencies” between performance metrics and ordering behaviors, J&J’s CareAdvantage offering helped Spectrum Health cut weekly out-of-stock devices by 49 percent. Moreover, the program enabled the hospital network to reduce expedited shipping fees a staggering 96 percent and the average number of stock-less days by 18 percent.
“Johnson & Johnson uses its computing power to pull data, run reports, and give recommendations on changing a particular part number. Some part numbers had not been looked at for a while and we were using more supplies and running out as the delivery system grew. We never looked back to see,” Kurt Knoth, Spectrum Health system supply chain vice president, told the New York, N.Y.-based trade journal Health Data Management in August. “There is so much opportunity out there that just gets hidden. Every dollar saved through the supply chain means less worrying about how to pay for doctors and nurses, or cutting people or supplies, while better supporting clinicians.”
And better clinician support ultimately leads to better patient outcomes, a key driver of business model innovation within the value-driven healthcare sphere. In many cases, these strategies are expanding beyond products to include devices and services that reward value rather than volume and meet increasing payer, provider, patient, and caregiver demands.
Some companies are building new business models around data and analytics in an effort to demonstrate value to payers and influence patient behavior. J&J’s 10-month-old CareAdvantage initiative, for instance, analyzes data, identifies mutual areas of focus, and clearly defines goals and responsibilities to improve supply chain optimization. The program helped Salt Lake City, Utah-based Intermountain Healthcare increase inventory returns by 25 percent, save $17,000 annually in product costs, and reduce stockouts 40 percent. “When you’re spending all your time talking about inventory, it’s hard to get to what truly matters,” declared Gordon Slade, supply chain logistics director for the 22-hospital Intermountain network. “Improving outcomes and quality of life—those are the conversations we want to be having, rather than, ‘we might be out of stock.’ “
J&J launched the CareAdvantage service in January, touting it as a “holistic, data-driven approach, grounded in insights” to help companies shift to alternative payment models tied to quality or value. The Advanced Medical Technology Association (AdvaMed) followed suit four months later, launching a new initiative called “Value Framework” to help medtech firms formulate value-based arguments.
AdvaMed’s program features extensive tables of questions, diagnostic technology-specific tools, and reports on understanding evidence and use cases. The framework also allows for different patient populations and urges device companies to contemplate product value over various timeframes (i.e., short- and long-term).
Under development for roughly two years with Deloitte Consulting LLP, the framework incorporates four key “value drivers” critical to understanding any assessment of technology:
Clinical impact: The extent of clinical utility and health outcomes associated with a medical technology.
Non-clinical patient impact: The impact on non-medical benefits for the patient (or caregiver) such as patient experience and outcomes (e.g., out-of-pocket costs).
Care delivery revenue and cost impact: A technology’s impact on revenue or cost for a provider or payer via financial incentives associated with care quality metrics, as well as impact on clinical workflow and other sources of operating efficiency.
Public/population impact: A technology’s impact on the overall healthcare system and employers, or society as a whole.
“These value drivers go beyond traditional clinical efficiency to capture economic impact across all parts of the healthcare ecosystem, as well as newer patient-focused considerations,” explained Mary Cummins, principal, Deloitte Consulting, life sciences and healthcare strategy practice. “The drivers take into account how a medical technology can impact care delivery and enable meaningful improvements in effectiveness and efficiency that providers and payers need.”
Besides helping medtech firms create comprehensive value-based arguments for existing devices, AdvaMed’s framework encourages manufacturers to integrate value-based thinking into the earliest stages of product development, from ideation and design to evidence collection and regulatory approval.
Whether broad or narrow in scope, medtech business models that embrace value-based care are fundamentally different from traditional fee-for-service contracts, as they require up-front investments and risk sharing. The plans themselves vary greatly, with some companies forging broad, multi-year partnerships that help payers and providers solve difficult challenges. Others are more product-centric but significantly change the way device firms are paid by linking reimbursement to demonstrated outcomes.
Medtronic plc chose the latter strategy, releasing a value-priced knee implant tied to the new bundled payment model developed by the Centers for Medicare and Medicaid Services (CMS). Debuting in the first half of 2017, the company’s Responsive Orthopedics Total Knee Arthroplasty System features streamlined surgical tools, a symmetric poly design, a locking mechanism for improved anatomical matching, and various surfaces (cruciate retaining, posterior stabilized, ultra conforming, medial pivot). Medtronic acquired the technology through its 2016 purchase of Minneapolis, Minn.-based Responsive Orthopedics, a niche maker of lower-cost artificial joints.
“Medtronic is here to help speed the adoption of value-based healthcare in orthopedics by helping hospitals drive down costs while keeping outcomes top of mind,” Geoff Martha, executive vice president of the multinational’s Restorative Therapies Group, said in a news release. “This is about more than just offering implants or individual technologies and services; it’s about partnering with all stakeholders throughout the entire episode of care to enable patient-centered care at the best value.”
J&J extended its value-based stakeholder partnerships beyond its CareAdvantage program with the formation of a professional education institute comprising 26 facilities and a comprehensive network of online education and collaborations across various specialties. The institute’s curriculum includes on-site classes, virtual reality, and app-based surgical simulation training to help clinical and non-clinical healthcare professionals improve outcomes, increase patient satisfaction, and reduce costs.
“Around the world, significant disparities in healthcare professional education and training, coupled with a shortage of health workers, means that some patients and consumers are unable to benefit from the latest medical knowledge and expertise,” Sandra Humbles, vice president of Global Education Solutions for Johnson & Johnson Medical Devices Companies, noted in announcing the institute’s formation in early November. “Globally, healthcare systems face resource challenges, increasing numbers of patients that lack access to quality healthcare and the continuous need for professional training on new technologies and procedures. Increasing access to healthcare education to improve outcomes and enhance the patient experience—while also reducing costs—is crucial. The Johnson & Johnson Institute will harness the breadth, reach, resources, and collective passion of the Johnson & Johnson Family of Companies to address these needs.”
Read more: http://bit.ly/yir1703
MDUFA 4.0
It was an unusual bill signing, to say the least.
There were no photo opps, no Oval Office guest lists, no special pens, and no speeches. Most surprisingly, there were no theatrics: Not one narcissistic remark was made, or self-serving tweet was sent.
Really.
It was, for all intents and purposes, a rare display of reclusion by President Donald J. Trump.
Thus marked the sequestered birth of the much-debated, long-awaited FDA Reauthorization Act of 2017 (FDARA), signed into law quietly and unceremoniously on Aug. 18. The Trump-approved legislation includes the sixth rendition of prescription drug user fees, the fourth iteration of medical device user fees, and the second versions of both the biosimilar and generic drug user fee agreements.
Capping two years of U.S. Food and Drug Administration (FDA) negotiations with industry and stakeholders, FDARA sets user fees through fiscal 2022 and finances roughly 60 percent of the agency’s pre-market review costs. Under the law’s Medical Device User Fee Amendments (MDUFA IV), FDA is authorized to collect $183.3 million in FY18 fees, a 30 percent increase from the previous year’s post-adjustment estimate.
“FDARA builds upon the goals outlined in previous user fee agreements and in the 21st Century Cures Act and will help us continue the essential work we are doing in many of our priority areas,” directors of the FDA’s drug, device, and biologics centers wrote in an Aug. 21 blog on the agency’s website. “The new law provides critical support for important FDA activities related to medical product regulation.”
Among the support provided by MDUFA IV include enabling FDA to inspect medical device facilities based on risk, and establishing a flexible, more efficient path to market for certain product accessories. The risk-based inspections, according to FDA officials, will allow the agency’s Center for Devices and Radiological Health to better focus its resources while providing greater predictability and transparency to the inspection process.
MDUFA IV also sets performance goals for various regulatory sanctions, committing for the first time to deciding half of all fiscal 2018 de novo requests within 150 days of filing, and resolving pre-submission meeting applications within two weeks. In addition, the agency agreed to provide written feedback for pre-submissions within 70 days or five days before a scheduled meeting for at least 1,530 pre-submissions received in FY18.
For 510(k) applications, FDA aims to decide 95 percent of 510(k) submissions within 90 days of filing and document an application’s problems after 100 days. MPA reviews have a similar timeline, requiring FDA to relay deficiencies to 95 percent of applicants within 60 days of filing and issue a decision for 90 percent of submissions not requiring an Advisory Committee review within 180 days.
“The MDUFA IV agreement...will expedite the availability of innovative new products, and its enhancements will continue to increase the efficiency of FDA’s programs,” Jeffrey Shuren, M.D., J.D., director of FDA’s Center for Devices and Radiological Health, told a U.S. Senate committee in March. “Improvements in total time to decision, transparency, consistency, and predictability will benefit industry, healthcare providers, and most importantly, patients.”
And FDA as well. MDUFA IV raises user fees for all regulatory submissions, with most averaging a 33 percent hike. The law, which took effect Oct. 1, softens the blow a bit for small businesses (less than $100 million in sales), though it doesn’t exempt them from a 37 percent increase in FDA Establishment fees, or lower any prices as in years past.
Nevertheless, the most significant MDUFA IV fees and hikes are ascribed to FDA’s 510(k) applications and de novo reviews. The agreement more than doubles 510(k) fees for moderate and large-size companies ($4,690 to $10,566) and boosts application costs 13 percent for small firms ($2,345 to $2,642).
The pecuniary onus is even greater for de novo reviews, as the law establishes first-time charges of $23,307 for small companies and a staggering $93,229 for moderate/large entities. Pediatric device submissions are exempt from the fees, as are uncommercialized state- or federal government-driven innovations. The law also waives fees for companies providing FDA with additional data on previously submitted de novo filings.
Resubmissions, however, are subject to additional user fees based on the applicant’s new (or old) chosen market pathway, according to MDUFA IV.
In exchange for its costly de novo fees, FDA promises to improve review times over the next five years, aiming to provide a final decision with 150 days for 70 percent of de novo requests submitted in FY2022. The goal seems practical, considering the amount of work involved with de novo submissions, and FDA’s perpetual failure to meet the statutorily-defined 120-day review period.
De novo review times have fallen significantly since 2009, but the process is still slow and unpredictable, averaging about 259 days, according to FDA data. Further reductions, though, could provide companies with a better, more predictable market pathway and consequently justify the new fees.
“The de novo pathway is essential for allowing brand technology to be incorporated into the whole 510(k) process because otherwise, they’d all have to go PMA, which is completely unrealistic and would kill a lot of technologies,” medtech law attorney Jeffrey K. Shapiro of Washington, D.C., told Bloomberg BNA in June. “I think that while the user fees are going to be a burden on industry, particularly small companies, there will be a trade-off in terms of greater certainty and a reduction in the timeline and greater predictability. I think investors are going to be happier factoring in paying the fees and getting that certainty of time to be able to get to market.”
Perhaps, but not all companies are willing to make such concessions. Healthcare attorney Bradley Merrill Thompson contends the high fees could make the PMA process more appealing to larger companies and discourage financially strapped smaller firms from developing “groundbreaking innovations.”
“The bottom line is I started to see some hope for the process,” he noted to Bloomberg BNA, “but the amount of the user fee I fear will discourage innovation.”
Michael Drues agrees.
“I’m not against having a de novo user fee, but the numbers concern me quite a bit,” said Drues, a Ph.D. and president of Grafton, Mass.-based Vascular Sciences, an education, training, and consulting company offering a range of services to medical device, pharmaceutical, and biotech firms. “Comparing the de novo fee to a 510(k), the de novo fee is 10 times more. The question is why? To be fair, a de novo is a little more work for the FDA—a new regulation and product code have to be created, and in some cases, special controls have to be created. But is it 10 times more work for the FDA than a 510(k)? Absolutely not. I think if we are going to have de novo user fees they should be comparable or perhaps slightly higher than a 510(k) but certainly not a 10-fold increase. Can you think of any better way to stifle or kill innovation than by creating those kinds of user fees for the de novo?”
No better way at all.
Read more: http://bit.ly/yir1704
If at First You Don’t Succeed...Fail, Fail Again
The headlines read like a comedy of errors.
“Obamacare Repeal Might Have Just Died Tonight,” New York magazine reported in early January.
Five months later, the magazine ran stories within days of each other on stalled repeal efforts and past strategies for killing the controversial healthcare law. Its long- awaited death seemed close, if not imminent, according to news accounts.
Then, damnation— “Obamacare Lives.” (July 28)
Followed by a confirmation: “Obamacare is Officially Not Collapsing.” (Aug. 24)
Curses! Time to move on.
Or not: “Hold On—Could the GOP’s Last-Ditch Effort to Kill Obamacare Actually Pass?” (Sept. 15)
Nope. False alarm. “The GOP’s Latest Attempt to Repeal Obamacare Won’t Even Get a Vote.” (Sept. 26)
A frustrated Donald Trump was willing to work with Democrats on fixing Obamacare until House Speaker Paul Ryan nixed the idea in the fall. ”Ryan Rejects Bipartisan Health-Care ‘Fix,’ and Now Trump Does Too.” (Oct. 18)
But why listen to Ryan? “Why You Might be Seeing Bipartisan Health-Care Bill in December.” (Oct. 19)
Indeed, Obamacare—a.k.a., the 2010 Affordable Care Act (ACA)—led a wildly chaotic existence this year as President Trump and his Republican cohorts worked to unravel President Barack Obama’s signature domestic achievement. Their tactics changed as often as Trump’s version of the truth, angering Democrats, confounding constituents, and ultimately spawning an independent battle plan from the commander-in-chief himself.
Trump’s solo campaign against Obamacare entailed scrapping health insurance subsidies for low-income families and authorizing the sales of cheaper policies with fewer benefits and fewer consumer protections. The strategy rattled America’s healthcare and political worlds, as it threatened to boost premiums, disrupt insurance markets, and shove Republicans into a renewed civil war over the ACA.
U.S. Senate Health Committee Chairman Lamar Alexander (R-Tenn.) tried heading off hostilities via a bipartisan solution he negotiated with U.S. Sen. Patty Murray (D-Wash.) that would fund cost-sharing insurer subsidies for two years and give states more flexibility to waive Obamacare rules. The proposed legislation also would let more people sign up for high-deductible healthcare insurance, encourage the sale of policies across state lines, and streamline the process of obtaining federal regulatory waivers for innovative health insurance plans.
Perhaps most importantly, the changes offered under the Alexander-Murray compromise upheld protections for patients with pre-existing conditions and maintained coverage for essential health services like maternity care and cancer treatment. The untitled bill had garnered 24 co-sponsors as of Oct. 19—12 from each political party.
“Every Republican in the House of Representatives who voted to repeal and replace Obamacare this year voted for a provision that continued the cost-sharing payments for two years,” Alexander said. “Our bill does the same thing.”
Maybe so, but it nevertheless faces an uphill battle for support: Senate Finance Committee Chairman Orrin G. Hatch wasn’t impressed with the bill (“It’s more of the same. More big spending, more big government, more lack of a solution,” the Utah Republican told CQ Roll Call), and U.S. Rep. Mark Walker (R-N.C.), dissed the deal on social media.
“The GOP should focus on repealing and replacing ObamaCare, not on trying to save it,” the Republican Study Committee chairman wrote on Twitter. “This bailout is unacceptable.”
President Trump thought so too.
“I am supportive of Lamar as a person & also of the process, but I can never support bailing out ins co’s who have made a fortune w/O’Care,” the president tweeted on Oct. 18.
Missing in all of Trump’s ACA twitter noise, however, was his thoughts on the 2.3 percent medical device tax. The president—and Congress, for the most part—were unusually tight-lipped about the levy this year, leaving the medtech industry staring down old demons as the final weeks of suspension drew to a close.
Created in 2010 to help fund expanded healthcare coverage under the ACA, the device tax was suspended for two years in late 2015 as part of a compromise $1.8 trillion spending package. The levy is scheduled to be reinstated on Jan. 1 unless lawmakers act by year’s end to further suspend or repeal it—an outcome that is becoming increasingly unlikely considering the Trump administration’s multiple fumbles with replacing Obamacare.
“Congress must act now to permanently repeal the device tax. Otherwise, medical technology companies will be hit with a tax increase of $20 billion at the end of the year, which will create a ripple effect for years to come,” Todd Usen, president of the Medical Systems Group at Olympus Corp. of the Americas, wrote in a late-summer op-ed column in the Philadelphia Inquirer. “As a global health-care company focused on minimally invasive medical solutions, we know firsthand the medical device tax will disrupt the industry’s ability to make investments in research and development and bolster the long-term medical innovation pipeline. The industry is making budgetary and planning decisions now that will impact innovations in a decade and needs the certainty that would be engendered through full repeal.”
Industry leaders fought fervently against the tax since its 2013 implementation, arguing the levy decimated the funding necessary to drive innovation and fuel growth. The American Action Forum estimates the device tax cost the industry more than 28,800 jobs during its three-year lifecycle, and warns its resurgence could eliminate another 25,000 positions through 2021. Reinstatement also is likely to force some old habits: Eighty-eight percent of “innovators” plan to slow hiring and/or eliminate jobs upon the tax’s return, and 77 percent will either slow or stop expansion plans, according to a survey of more than 100 medtech companies by the Washington, D.C.-based Medical Device Manufacturers Association (MDMA). Additionally, 83 percent of the group’s survey respondents said they would decrease R&D investments if the tax took effect again.
“Medical technology innovators have already made difficult decisions as a result of the uncertainty over the device tax, and now is the time to put a permanent end to a policy that the overwhelming majority of Congress agrees is a bad idea,” MDMA President and CEO Mark Leahey said in formal statement in October. “There continues to be strong bipartisan support to repeal the medical device tax, but time is of the essence. It hovers like a dark cloud over innovation and job creation, and it is critical that Congress finishes the job.”
Start preparing for the rainstorm—just in case.
Read more: http://bit.ly/yir1706
Seeking Scale and Scope
Nothing is impossible in Jay Gatsby’s world.
The title character of F. Scott Fitzgerald’s 1925 literary classic believes he can achieve anything, regardless of its plausibility. At one point in the novel, for example, he boldly protests a friend’s misgivings about manipulating time and reuniting with a former lover.
“...she doesn’t understand,” Gatsby complains, noting the emotional distance he feels from his sweetheart. “She used to be able to understand. We’d sit for hours—”
“I wouldn’t ask too much of her,” Gatsby’s friend (and story narrator) Nick advises. “You can’t repeat the past.”
“Can’t repeat the past?” Gatsby cries incredulously. “Why of course you can!”
Such Gatsbyan musings briefly materialized earlier this year as medtech companies continued brokering M&A agreements that secured long-term growth through diversification and economies of scale. With a handful of sizable transactions announced in the first half of 2017 (among them, two mega-mergers), it seemed as if total deal value would easily match 2015’s unprecedented levels.
But, much like Gatsby’s futile attempts to resurrect history, the medtech industry fell far short in recapturing its own past magic, as the blockbuster deals that defined the 2014-2015 gilded era dried up in the second half of 2017.
Nevertheless, M&A activity accelerated considerably this year in the orthopedic space as Globus Medical, NuVasive Inc., and Stryker Corp. arbitrated acquisitions that strengthened and/or boosted their scale in increasingly competitive therapeutic areas. Globus Medical’s midsummer deal for Swiss robotics developer KB Medical, for instance, is expected to help shape future enhancements to the larger firm’s ExcelsiusGPS robotic guidance and navigation system. KB Medical’s AQrate robotic system is used during lumbar fusion procedures to plan operations and guide instrument positioning for pedicle screw placement during the minimally invasive approach. The company received a CE mark for the AQrate system last year after completing a clinical trial treating more than 20 patients.
“The addition of KB Medical will enable Globus Medical to accelerate, enhance and expand our product portfolio in imaging, navigation and robotics,” president of Emerging Technologies Dave Demski said. “KB Medical’s team of technology development professionals, its strong IP portfolio and shared philosophy for robotic solutions in medicine strengthens Globus Medical’s position in this strategic area.”
Likewise, NuVasive’s purchase of Vertera Spine gives the company sole bragging rights to both polyetheretherketone (PEEK) and titanium porous interbody devices. Vertera’s biomaterial (Scoria) is grown from solid PEEK material, creating a seamless surface-to-solid material interface that is reportedly more durable than metal coatings and twice as strong as trabecular bone under shear loading.
Besides enhancing its PEEK technology offering, the Vertera deal also improves NuVasive’s chances of achieving its $1 billion revenue goal this year. The company moved well within range of its target with last year’s purchases of Ellipse Technologies, Biotronic NeuroNetwork, and LessRay software.
Better scale prompted other deals as well this year, enabling Marietta, Ga.-based Amendia to bolster its pedicle screw system offering and cervical product lines through Spinal Elements; Integra LifeSciences to expand its footprint in advanced wound care through Derma Sciences ($204 million); DePuy Synthes to accelerate spine market growth through the acquisition of Interventional Spine’s expandable cage technology; ConMed to strengthen its arthroscopy portfolio through the purchase of MedShape’s ExoShape Anterior Cruciate Ligament fixation system; and Stryker to fortify its advanced fluorescence imaging technology and minimally invasive surgical solutions for spine trauma via NOVADAQ Technologies Inc. ($701 million) and VEXIM ($216 million). Moreover, K2M Group Holdings Inc. padded its cervical spine showing by purchasing the PALO ALTO Cervical Static Corpectomy Cage System from Cardinal Spine, and enhanced its expandable interbody and 3D-printed product lineups through a patent portfolio agreement.
“...medtechs recognize that acquisitions that build end-to-end capabilities in a particular therapeutic area or expand the company’s geographic or technological reach are one of the fastest paths to growth,” EY analysts state in the firm’s “Pulse of the Industry 2017“ report.
But they’re not the only path to growth. Diversification inspired numerous transactions this year, pairing Avalign Technologies with Millennium Surgical and Thortex; Smith and Nephew plc with Rotation Medical Inc.; and DePuy Synthes with both Sentio LLC and Tissue Regeneration Systems. The latter deal gives DePuy the ability to develop personalized healthcare solutions in Trauma that potentially can help improve both patient satisfaction and clinical outcomes—key bellwethers in the migration toward value-based care.
Divestitures and spin-outs led to a few pairings this year too, most notably Johnson & Johnson and Integra LifeSciences. New Brunswick, N.J.-based J&J agreed in February to sell its Codman Neurosurgery business (sans the neurovascular and drug delivery divisions) for $1.05 billion to Integra LifeSciences, which has recently been restructuring its portfolio to build strong platforms in core areas like orthopedics and tissue technologies. J&J, meanwhile, has executed a similar strategy in its Medical Devices business, discarding low-performing segments to focus on higher growth markets such as orthopedics, surgery, and vision care.
“Divestitures and spin-outs allow medtechs to capture additional value by improving capital efficiency, reducing operational complexity, and reallocating capital to higher-growth businesses as the industry invests more R&D dollars in the development of innovative products that demonstrate value in an era of price pressures,” the EY report states. “Indeed...Johnson & Johnson’s divestiture of Codman Neurosurgery to Integra LifeSciences suggests capital efficiency and portfolio optimization will be an ongoing trend in 2018.”
Time will tell.
Read more: http://bit.ly/yir1708
Legal Troubles for DePuy
In the videos, they’re all smiling. Each and every one. There’s Rebecca, beaming broadly while walking hand-in-hand with two grandchildren; Roger, chuckling softly to his wife of 50 years; and Laura, laughing boldly and heartily during a trike motorcycle outing.
There’s also Bob, contentedly playing handball; Pam, cheerfully fixing a clothing store display; and Lani, grinning ever so slightly on horseback.
Absent from this sanguine sextet is Rocky Thompson of Pitkin, La., an unincorporated community of merely 576 residents. Thompson is not smiling. Nor is he happy.
Thompson could have been as happy as his counterparts: He replaced his ailing left knee with DePuy Synthes’ Attune Knee System, just like the others. But his recovery and post-replacement life has been markedly different than the merry band of Attune patient ambassadors featured on DePuy Synthes’ website.
Thompson blames the Attune system for his poor quality of life, claiming in a federal lawsuit that the product is defective and “unreasonably dangerous.” The suit, filed Sept. 26 in Louisiana district court, is reportedly the first to name Attune.
In the 43-page complaint, Thompson indicates he underwent knee replacement surgery in January 2015, swapping his natural joint for an Attune system comprised of a fixed tibial insert and fixed tibial baseplate. After the procedure, however, Thompson began experiencing “severe and persistent pain, discomfort, instability and difficulty ambulating” due to aseptic loosening and a tibial baseplate defect, according to the lawsuit. He underwent revision surgery in July this year—18 months after receiving the Attune joint.
Thompson’s lawsuit attributes the Attune’s defect to mechanical loosening, which is typically caused by a weakened or failing bond between the tibial baseplate and implant-cement surface. When such loosening occurs, the artificial knee can detach from existing bone, resulting in failure.
When it debuted in 2014, the Attune knee was considered an “advancement” in knee replacement options due to its S-curve design, gradual radius reduction, and a tibial base that optimizes kinematics while reducing wear. The technology was based on six years of research and inspired by overall high rates of patient dissatisfaction with artificial knees (up to 20 percent, industry-wide). In releasing Attune, DePuy promised patients greater functional benefits and a better range of motion compared to other implants.
Thompson, however, believes the product did more harm than good. “...in reality, the Attune device did not deliver on these promises, resulting in significantly higher failure rates than previous DePuy knee counterparts due to the debonding of the tibial baseplate,” his lawsuit states. “As a result, thousands of knee replacement patients implanted with Attune devices have had more expensive, more dangerous and less effective Total Knee Replacement surgeries, and many have required or will require expensive and dangerous knee revision surgery to remove and replace the defective Attune device.”
Although DePuy hasn’t responded directly to Thompson’s lawsuit, the company has released volumes of Attune study data confirming low revision rates and high implant survivorship. Statistics from the National Joint Registry for England, Wales, Northern Ireland and the Isle of Man (NJR), for example, show an estimated cumulative revision rate of 1.3 percent at four years (98.7 percent implant survivorship) for 10,605 Attune knees, which compares favorably to the 1.9 percent cumulative revision rate (98.1 percent implant survivorship) for the overall class of knee replacements.
The numbers are even better Down Under: The Attune Cruciate Retaining Knee boasts a 2.1 percent cumulative revision rate at three years, which is consistent with the class, while the Posterior Stabilized Knee exhibited a 1.1 percent rate at three years, which compares favorably to the class, according to data from 8,3841 Attune knees tracked by the Australian Orthopaedic Association National Joint Replacement Registry. Attune also scored well in New Zealand, claiming an overall 0.66 revision rate per 100 component years, with 33 revisions (of 3,795 implants), data from an 18-year joint registry report show.
The numbers are solid in Michigan as well, with the Attune knee showing a 2.42 percent revision rate after three years based on 4,870 implants from 2012 to 2016, the Michigan Arthroplasty Registry Collaborative Quality Initiative (MARQI) concluded.
Furthermore, a radiostereometric (RSA) analysis conducted by the Canadian RSA Network found the Attune tibial base to be stable, moving only 0.17 mm between one and two years. The results are consistent with implants that have acceptable revision rates due to aseptic loosening. A second, randomized-controlled RSA study conducted in the Netherlands compared Attune implant migration to patients implanted with SIGMA knees and reported no difference in implant migration.
“Attune has been in clinical use for over six years now, and we have over half a million cases [of it used] around the world,” Rajit Kamal, global knee platform leader for DePuy Synthes, told Orthopedic Design & Technology. “There’s a solid body of evidence that very clearly shows that Attune is delivering on the intended objectives we had. It is showing better patient-reported outcomes, patient satisfaction, and implant survivorship in-line with the class.”
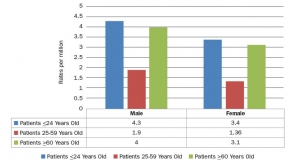
Maybe so, but a growing body of evidence suggests otherwise. Thompson’s lawsuit cites 1,400 reports of Attune knee failures from the U.S. Food and Drug Administration’s (FDA) MAUDE (Manufacturer and User Facility Device Experience) database, with roughly 633 of those failures resulting in revision surgeries. The suit, however, does not specify a reason for either the failures or the revisions.
Similarly, an April 2017 study published in the Journal of Knee Surgery shows a “high” rate of Attune tibial loosening at the implant-cement interface. Researchers identified 15 cases from three hospitals and referenced 21 additional reports of tibial loosening in the MAUDE database within a two-month period as well as “numerous other tibial failures” also reported without the mechanism for failure specified.
DePuy, of course, questioned the study’s accuracy, noting the absence of a rate calculation and radiographic technique or positioning. The company also took issue with the researchers’ use of MAUDE database statistics, claiming it conflicts with the intent of the database and FDA guidance. “The FDA’s guidance is that, ‘MDR data alone cannot be used to establish rates of events, evaluate a change in event rates over time or compare event rates between devices. The number of reports cannot be interpreted or used in isolation to reach conclusions about the existence, severity, or frequency of problems associated with devices,’ “DePuy said in a formal response to the Journal of Knee Surgery study.
The company’s defense resonated with Cleveland Clinic doctor Michael A. Mont, who criticized the study for its lack of a denominator. “When one looks at a case series, we really do not know the denominator, and this certainly could have been a series of cases out of more than 100,000 done, which would make this a rare event,” Mont wrote in a Journal opinion piece. “Therefore, simply publishing cases is not evidence-based medicine, as we do not know how many of these were implanted. For example, ceramic fractures occur very infrequently, maybe one in 10,000; and yet, a small series could easily be published. The key is to know the denominator. Although this report may raise some interest, I would like to think that it is only an isolated group of failures that can occur with any device in the field from any manufacturer.”
Perhaps, but Thompson disagrees. He contends the Attune knee’s tibial base plate is defective, and points to DePuy’s newest iteration, the Attune S+ as proof. “Defendants’ research, design, marketing, and placement of the Attune S+ with new design features on the market aimed at increasing fixation is an admission that the original Attune tibial base plate was defective in its composition and/or construction,” he argues in his lawsuit.
DePuy, though, dismisses such allegations, asserting the Attune S+ is designed to further improve the industry-wide issue of aseptic loosening rates. The S+ technology, in development for the last two years, combines macrolock features and a microblast finish to inhibit lipid infiltration on the tibial base surface and increase pull-off strength under common simulated intra-operative surgical conditions. The product’s rougher surface is intended to increase cement-implant interdigitation and reduce the amount of fluids that might bleed into the cement-implant interface. In addition, four cement pockets with 45-degree undercuts and macro geometry aim to improve mechanical fixation with cured cement.
“Based on the literature out there, benchtop testing, and cadaveric studies, we found that the biggest variable that causes a reduction of the strength of fixation between the inner part [of the implant] and the cement is the introduction of lipids or marrow between those two components (cement and implant),” Liam Rowley, worldwide product director, Knees, for DePuy Synthes, noted to ODT. “We found that a combination of a macrolock plus an optimized microblast—the combination of those two together—makes the product much more resistant to lipids infiltration. So far, the Attune S+ has outperformed everything we’ve measured against in terms of benchtop testing. We’re really excited about it, because we think we’ve got a product that will really raise the bar for the whole industry in terms of tibial fixation.”
Read more: http://bit.ly/yir1711
The medtech industry showed steely resolve this year in the wake of political chaos and meteorological mayhem, shortening supply chains and streamlining operations to weather the impacts of a tyrannical hurricane trifecta, and maintaining the fight for full repeal of the 2.3 percent medical device tax.
That battle proved challenging, as the controversial levy became lost among a litany of woes endangering President Donald Trump’s legislative agenda, among them: multiple investigations into Russian interference in the 2016 election; former FBI Director James Comey’s controversial firing; staff infighting; the ban on transgender military personnel; high-profile resignations; the president’s word war with North Korean leader Kim Jong-un; and vacillating tensions with Congress.
Despite all the turmoil, the industry never strayed from its goal, though it did eventually adjust its expectations. “There are opportunities—fewer and fewer though to get full repeal,” Clayton Hall, vice president of government affairs for the Medical Device Manufacturers Association, told a Medmarc Insurance Group webinar audience in August. “The likeliest outcome right now is probably an additional suspension. But there are some opportunities to get full repeal. And that’s what we’re fighting for.”
Medtech’s mettle also manifested itself in the sector’s sustained progression toward value-based care, improved payer engagement, data analytics, and digital health. The U.S. Food and Drug Administration (FDA) helped champion the latter cause in September with a first-of-its kind program designed to revolutionize digital health regulation in the United States. The FDA’s Pre-cert pilot program is intended to develop an approach toward digital health technology by looking at the software developer or digital health technology developer, rather than primarily at the product. Participants in the FDA’s precertification pilot program (FDA Pre-cert) include Apple, Fitbit, Johnson & Johnson, Pear Therapeutics, Phosphorus, Roche, Samsung, Tidepool, and Verily.
“Our method for regulating digital health products must recognize the unique and iterative characteristics of these products,” FDA Commissioner Scott Gottlieb said in launching the program. “We need to modernize our regulatory framework so that it matches the kind of innovation we’re being asked to evaluate, and helps foster beneficial technology while ensuring that consumers have access to high-quality, safe and effective digital health devices.”
Besides propelling its assimilation of digital health in 2017, the device industry’s resolve also brought resolution to a number of longstanding issues. Zimmer Biomet Holdings Inc. and Orthofix International N.V., for example, put various federal investigations behind them, while Exactech ended its 32-year run as a public company. And just in the nick of time, the president signed a new medical device user fee agreement into law, capping two years of often tense negotiations between the industry and FDA.
Insight into these and other defining moments of 2017 can be gleaned from Orthopedic Design & Technology’s annual trip down memory lane. Enjoy the journey.
Less Volume, More Value
Spectrum Health is finding it hard to let go of tradition.
Despite all its innovations in telehealth and digital medicine, the non-profit healthcare system in Western Michigan still manages its supply chain the old-fashioned way—with paper and pencil.
Not surprisingly, such an archaic method has spawned more than its fair share of old-fashioned headaches, namely, keeping abreast of product supply levels and value. Earlier this year, however, the 12-hospital network began modernizing its supply chain management program through a Johnson & Johnson initiative designed to help health systems navigate value-based care.
Using data analytics to identify “critical interdependencies” between performance metrics and ordering behaviors, J&J’s CareAdvantage offering helped Spectrum Health cut weekly out-of-stock devices by 49 percent. Moreover, the program enabled the hospital network to reduce expedited shipping fees a staggering 96 percent and the average number of stock-less days by 18 percent.
“Johnson & Johnson uses its computing power to pull data, run reports, and give recommendations on changing a particular part number. Some part numbers had not been looked at for a while and we were using more supplies and running out as the delivery system grew. We never looked back to see,” Kurt Knoth, Spectrum Health system supply chain vice president, told the New York, N.Y.-based trade journal Health Data Management in August. “There is so much opportunity out there that just gets hidden. Every dollar saved through the supply chain means less worrying about how to pay for doctors and nurses, or cutting people or supplies, while better supporting clinicians.”
And better clinician support ultimately leads to better patient outcomes, a key driver of business model innovation within the value-driven healthcare sphere. In many cases, these strategies are expanding beyond products to include devices and services that reward value rather than volume and meet increasing payer, provider, patient, and caregiver demands.
Some companies are building new business models around data and analytics in an effort to demonstrate value to payers and influence patient behavior. J&J’s 10-month-old CareAdvantage initiative, for instance, analyzes data, identifies mutual areas of focus, and clearly defines goals and responsibilities to improve supply chain optimization. The program helped Salt Lake City, Utah-based Intermountain Healthcare increase inventory returns by 25 percent, save $17,000 annually in product costs, and reduce stockouts 40 percent. “When you’re spending all your time talking about inventory, it’s hard to get to what truly matters,” declared Gordon Slade, supply chain logistics director for the 22-hospital Intermountain network. “Improving outcomes and quality of life—those are the conversations we want to be having, rather than, ‘we might be out of stock.’ “
J&J launched the CareAdvantage service in January, touting it as a “holistic, data-driven approach, grounded in insights” to help companies shift to alternative payment models tied to quality or value. The Advanced Medical Technology Association (AdvaMed) followed suit four months later, launching a new initiative called “Value Framework” to help medtech firms formulate value-based arguments.
AdvaMed’s program features extensive tables of questions, diagnostic technology-specific tools, and reports on understanding evidence and use cases. The framework also allows for different patient populations and urges device companies to contemplate product value over various timeframes (i.e., short- and long-term).
Under development for roughly two years with Deloitte Consulting LLP, the framework incorporates four key “value drivers” critical to understanding any assessment of technology:
Clinical impact: The extent of clinical utility and health outcomes associated with a medical technology.
Non-clinical patient impact: The impact on non-medical benefits for the patient (or caregiver) such as patient experience and outcomes (e.g., out-of-pocket costs).
Care delivery revenue and cost impact: A technology’s impact on revenue or cost for a provider or payer via financial incentives associated with care quality metrics, as well as impact on clinical workflow and other sources of operating efficiency.
Public/population impact: A technology’s impact on the overall healthcare system and employers, or society as a whole.
“These value drivers go beyond traditional clinical efficiency to capture economic impact across all parts of the healthcare ecosystem, as well as newer patient-focused considerations,” explained Mary Cummins, principal, Deloitte Consulting, life sciences and healthcare strategy practice. “The drivers take into account how a medical technology can impact care delivery and enable meaningful improvements in effectiveness and efficiency that providers and payers need.”
Besides helping medtech firms create comprehensive value-based arguments for existing devices, AdvaMed’s framework encourages manufacturers to integrate value-based thinking into the earliest stages of product development, from ideation and design to evidence collection and regulatory approval.
Whether broad or narrow in scope, medtech business models that embrace value-based care are fundamentally different from traditional fee-for-service contracts, as they require up-front investments and risk sharing. The plans themselves vary greatly, with some companies forging broad, multi-year partnerships that help payers and providers solve difficult challenges. Others are more product-centric but significantly change the way device firms are paid by linking reimbursement to demonstrated outcomes.
Medtronic plc chose the latter strategy, releasing a value-priced knee implant tied to the new bundled payment model developed by the Centers for Medicare and Medicaid Services (CMS). Debuting in the first half of 2017, the company’s Responsive Orthopedics Total Knee Arthroplasty System features streamlined surgical tools, a symmetric poly design, a locking mechanism for improved anatomical matching, and various surfaces (cruciate retaining, posterior stabilized, ultra conforming, medial pivot). Medtronic acquired the technology through its 2016 purchase of Minneapolis, Minn.-based Responsive Orthopedics, a niche maker of lower-cost artificial joints.
“Medtronic is here to help speed the adoption of value-based healthcare in orthopedics by helping hospitals drive down costs while keeping outcomes top of mind,” Geoff Martha, executive vice president of the multinational’s Restorative Therapies Group, said in a news release. “This is about more than just offering implants or individual technologies and services; it’s about partnering with all stakeholders throughout the entire episode of care to enable patient-centered care at the best value.”
J&J extended its value-based stakeholder partnerships beyond its CareAdvantage program with the formation of a professional education institute comprising 26 facilities and a comprehensive network of online education and collaborations across various specialties. The institute’s curriculum includes on-site classes, virtual reality, and app-based surgical simulation training to help clinical and non-clinical healthcare professionals improve outcomes, increase patient satisfaction, and reduce costs.
“Around the world, significant disparities in healthcare professional education and training, coupled with a shortage of health workers, means that some patients and consumers are unable to benefit from the latest medical knowledge and expertise,” Sandra Humbles, vice president of Global Education Solutions for Johnson & Johnson Medical Devices Companies, noted in announcing the institute’s formation in early November. “Globally, healthcare systems face resource challenges, increasing numbers of patients that lack access to quality healthcare and the continuous need for professional training on new technologies and procedures. Increasing access to healthcare education to improve outcomes and enhance the patient experience—while also reducing costs—is crucial. The Johnson & Johnson Institute will harness the breadth, reach, resources, and collective passion of the Johnson & Johnson Family of Companies to address these needs.”
Read more: http://bit.ly/yir1703
MDUFA 4.0
It was an unusual bill signing, to say the least.
There were no photo opps, no Oval Office guest lists, no special pens, and no speeches. Most surprisingly, there were no theatrics: Not one narcissistic remark was made, or self-serving tweet was sent.
Really.
It was, for all intents and purposes, a rare display of reclusion by President Donald J. Trump.
Thus marked the sequestered birth of the much-debated, long-awaited FDA Reauthorization Act of 2017 (FDARA), signed into law quietly and unceremoniously on Aug. 18. The Trump-approved legislation includes the sixth rendition of prescription drug user fees, the fourth iteration of medical device user fees, and the second versions of both the biosimilar and generic drug user fee agreements.
Capping two years of U.S. Food and Drug Administration (FDA) negotiations with industry and stakeholders, FDARA sets user fees through fiscal 2022 and finances roughly 60 percent of the agency’s pre-market review costs. Under the law’s Medical Device User Fee Amendments (MDUFA IV), FDA is authorized to collect $183.3 million in FY18 fees, a 30 percent increase from the previous year’s post-adjustment estimate.
“FDARA builds upon the goals outlined in previous user fee agreements and in the 21st Century Cures Act and will help us continue the essential work we are doing in many of our priority areas,” directors of the FDA’s drug, device, and biologics centers wrote in an Aug. 21 blog on the agency’s website. “The new law provides critical support for important FDA activities related to medical product regulation.”
Among the support provided by MDUFA IV include enabling FDA to inspect medical device facilities based on risk, and establishing a flexible, more efficient path to market for certain product accessories. The risk-based inspections, according to FDA officials, will allow the agency’s Center for Devices and Radiological Health to better focus its resources while providing greater predictability and transparency to the inspection process.
MDUFA IV also sets performance goals for various regulatory sanctions, committing for the first time to deciding half of all fiscal 2018 de novo requests within 150 days of filing, and resolving pre-submission meeting applications within two weeks. In addition, the agency agreed to provide written feedback for pre-submissions within 70 days or five days before a scheduled meeting for at least 1,530 pre-submissions received in FY18.
For 510(k) applications, FDA aims to decide 95 percent of 510(k) submissions within 90 days of filing and document an application’s problems after 100 days. MPA reviews have a similar timeline, requiring FDA to relay deficiencies to 95 percent of applicants within 60 days of filing and issue a decision for 90 percent of submissions not requiring an Advisory Committee review within 180 days.
“The MDUFA IV agreement...will expedite the availability of innovative new products, and its enhancements will continue to increase the efficiency of FDA’s programs,” Jeffrey Shuren, M.D., J.D., director of FDA’s Center for Devices and Radiological Health, told a U.S. Senate committee in March. “Improvements in total time to decision, transparency, consistency, and predictability will benefit industry, healthcare providers, and most importantly, patients.”
And FDA as well. MDUFA IV raises user fees for all regulatory submissions, with most averaging a 33 percent hike. The law, which took effect Oct. 1, softens the blow a bit for small businesses (less than $100 million in sales), though it doesn’t exempt them from a 37 percent increase in FDA Establishment fees, or lower any prices as in years past.
Nevertheless, the most significant MDUFA IV fees and hikes are ascribed to FDA’s 510(k) applications and de novo reviews. The agreement more than doubles 510(k) fees for moderate and large-size companies ($4,690 to $10,566) and boosts application costs 13 percent for small firms ($2,345 to $2,642).
The pecuniary onus is even greater for de novo reviews, as the law establishes first-time charges of $23,307 for small companies and a staggering $93,229 for moderate/large entities. Pediatric device submissions are exempt from the fees, as are uncommercialized state- or federal government-driven innovations. The law also waives fees for companies providing FDA with additional data on previously submitted de novo filings.
Resubmissions, however, are subject to additional user fees based on the applicant’s new (or old) chosen market pathway, according to MDUFA IV.
In exchange for its costly de novo fees, FDA promises to improve review times over the next five years, aiming to provide a final decision with 150 days for 70 percent of de novo requests submitted in FY2022. The goal seems practical, considering the amount of work involved with de novo submissions, and FDA’s perpetual failure to meet the statutorily-defined 120-day review period.
De novo review times have fallen significantly since 2009, but the process is still slow and unpredictable, averaging about 259 days, according to FDA data. Further reductions, though, could provide companies with a better, more predictable market pathway and consequently justify the new fees.
“The de novo pathway is essential for allowing brand technology to be incorporated into the whole 510(k) process because otherwise, they’d all have to go PMA, which is completely unrealistic and would kill a lot of technologies,” medtech law attorney Jeffrey K. Shapiro of Washington, D.C., told Bloomberg BNA in June. “I think that while the user fees are going to be a burden on industry, particularly small companies, there will be a trade-off in terms of greater certainty and a reduction in the timeline and greater predictability. I think investors are going to be happier factoring in paying the fees and getting that certainty of time to be able to get to market.”
Perhaps, but not all companies are willing to make such concessions. Healthcare attorney Bradley Merrill Thompson contends the high fees could make the PMA process more appealing to larger companies and discourage financially strapped smaller firms from developing “groundbreaking innovations.”
“The bottom line is I started to see some hope for the process,” he noted to Bloomberg BNA, “but the amount of the user fee I fear will discourage innovation.”
Michael Drues agrees.
“I’m not against having a de novo user fee, but the numbers concern me quite a bit,” said Drues, a Ph.D. and president of Grafton, Mass.-based Vascular Sciences, an education, training, and consulting company offering a range of services to medical device, pharmaceutical, and biotech firms. “Comparing the de novo fee to a 510(k), the de novo fee is 10 times more. The question is why? To be fair, a de novo is a little more work for the FDA—a new regulation and product code have to be created, and in some cases, special controls have to be created. But is it 10 times more work for the FDA than a 510(k)? Absolutely not. I think if we are going to have de novo user fees they should be comparable or perhaps slightly higher than a 510(k) but certainly not a 10-fold increase. Can you think of any better way to stifle or kill innovation than by creating those kinds of user fees for the de novo?”
No better way at all.
Read more: http://bit.ly/yir1704
If at First You Don’t Succeed...Fail, Fail Again
The headlines read like a comedy of errors.
“Obamacare Repeal Might Have Just Died Tonight,” New York magazine reported in early January.
Five months later, the magazine ran stories within days of each other on stalled repeal efforts and past strategies for killing the controversial healthcare law. Its long- awaited death seemed close, if not imminent, according to news accounts.
Then, damnation— “Obamacare Lives.” (July 28)
Followed by a confirmation: “Obamacare is Officially Not Collapsing.” (Aug. 24)
Curses! Time to move on.
Or not: “Hold On—Could the GOP’s Last-Ditch Effort to Kill Obamacare Actually Pass?” (Sept. 15)
Nope. False alarm. “The GOP’s Latest Attempt to Repeal Obamacare Won’t Even Get a Vote.” (Sept. 26)
A frustrated Donald Trump was willing to work with Democrats on fixing Obamacare until House Speaker Paul Ryan nixed the idea in the fall. ”Ryan Rejects Bipartisan Health-Care ‘Fix,’ and Now Trump Does Too.” (Oct. 18)
But why listen to Ryan? “Why You Might be Seeing Bipartisan Health-Care Bill in December.” (Oct. 19)
Indeed, Obamacare—a.k.a., the 2010 Affordable Care Act (ACA)—led a wildly chaotic existence this year as President Trump and his Republican cohorts worked to unravel President Barack Obama’s signature domestic achievement. Their tactics changed as often as Trump’s version of the truth, angering Democrats, confounding constituents, and ultimately spawning an independent battle plan from the commander-in-chief himself.
Trump’s solo campaign against Obamacare entailed scrapping health insurance subsidies for low-income families and authorizing the sales of cheaper policies with fewer benefits and fewer consumer protections. The strategy rattled America’s healthcare and political worlds, as it threatened to boost premiums, disrupt insurance markets, and shove Republicans into a renewed civil war over the ACA.
U.S. Senate Health Committee Chairman Lamar Alexander (R-Tenn.) tried heading off hostilities via a bipartisan solution he negotiated with U.S. Sen. Patty Murray (D-Wash.) that would fund cost-sharing insurer subsidies for two years and give states more flexibility to waive Obamacare rules. The proposed legislation also would let more people sign up for high-deductible healthcare insurance, encourage the sale of policies across state lines, and streamline the process of obtaining federal regulatory waivers for innovative health insurance plans.
Perhaps most importantly, the changes offered under the Alexander-Murray compromise upheld protections for patients with pre-existing conditions and maintained coverage for essential health services like maternity care and cancer treatment. The untitled bill had garnered 24 co-sponsors as of Oct. 19—12 from each political party.
“Every Republican in the House of Representatives who voted to repeal and replace Obamacare this year voted for a provision that continued the cost-sharing payments for two years,” Alexander said. “Our bill does the same thing.”
Maybe so, but it nevertheless faces an uphill battle for support: Senate Finance Committee Chairman Orrin G. Hatch wasn’t impressed with the bill (“It’s more of the same. More big spending, more big government, more lack of a solution,” the Utah Republican told CQ Roll Call), and U.S. Rep. Mark Walker (R-N.C.), dissed the deal on social media.
“The GOP should focus on repealing and replacing ObamaCare, not on trying to save it,” the Republican Study Committee chairman wrote on Twitter. “This bailout is unacceptable.”
President Trump thought so too.
“I am supportive of Lamar as a person & also of the process, but I can never support bailing out ins co’s who have made a fortune w/O’Care,” the president tweeted on Oct. 18.
Missing in all of Trump’s ACA twitter noise, however, was his thoughts on the 2.3 percent medical device tax. The president—and Congress, for the most part—were unusually tight-lipped about the levy this year, leaving the medtech industry staring down old demons as the final weeks of suspension drew to a close.
Created in 2010 to help fund expanded healthcare coverage under the ACA, the device tax was suspended for two years in late 2015 as part of a compromise $1.8 trillion spending package. The levy is scheduled to be reinstated on Jan. 1 unless lawmakers act by year’s end to further suspend or repeal it—an outcome that is becoming increasingly unlikely considering the Trump administration’s multiple fumbles with replacing Obamacare.
“Congress must act now to permanently repeal the device tax. Otherwise, medical technology companies will be hit with a tax increase of $20 billion at the end of the year, which will create a ripple effect for years to come,” Todd Usen, president of the Medical Systems Group at Olympus Corp. of the Americas, wrote in a late-summer op-ed column in the Philadelphia Inquirer. “As a global health-care company focused on minimally invasive medical solutions, we know firsthand the medical device tax will disrupt the industry’s ability to make investments in research and development and bolster the long-term medical innovation pipeline. The industry is making budgetary and planning decisions now that will impact innovations in a decade and needs the certainty that would be engendered through full repeal.”
Industry leaders fought fervently against the tax since its 2013 implementation, arguing the levy decimated the funding necessary to drive innovation and fuel growth. The American Action Forum estimates the device tax cost the industry more than 28,800 jobs during its three-year lifecycle, and warns its resurgence could eliminate another 25,000 positions through 2021. Reinstatement also is likely to force some old habits: Eighty-eight percent of “innovators” plan to slow hiring and/or eliminate jobs upon the tax’s return, and 77 percent will either slow or stop expansion plans, according to a survey of more than 100 medtech companies by the Washington, D.C.-based Medical Device Manufacturers Association (MDMA). Additionally, 83 percent of the group’s survey respondents said they would decrease R&D investments if the tax took effect again.
“Medical technology innovators have already made difficult decisions as a result of the uncertainty over the device tax, and now is the time to put a permanent end to a policy that the overwhelming majority of Congress agrees is a bad idea,” MDMA President and CEO Mark Leahey said in formal statement in October. “There continues to be strong bipartisan support to repeal the medical device tax, but time is of the essence. It hovers like a dark cloud over innovation and job creation, and it is critical that Congress finishes the job.”
Start preparing for the rainstorm—just in case.
Read more: http://bit.ly/yir1706
Seeking Scale and Scope
Nothing is impossible in Jay Gatsby’s world.
The title character of F. Scott Fitzgerald’s 1925 literary classic believes he can achieve anything, regardless of its plausibility. At one point in the novel, for example, he boldly protests a friend’s misgivings about manipulating time and reuniting with a former lover.
“...she doesn’t understand,” Gatsby complains, noting the emotional distance he feels from his sweetheart. “She used to be able to understand. We’d sit for hours—”
“I wouldn’t ask too much of her,” Gatsby’s friend (and story narrator) Nick advises. “You can’t repeat the past.”
“Can’t repeat the past?” Gatsby cries incredulously. “Why of course you can!”
Such Gatsbyan musings briefly materialized earlier this year as medtech companies continued brokering M&A agreements that secured long-term growth through diversification and economies of scale. With a handful of sizable transactions announced in the first half of 2017 (among them, two mega-mergers), it seemed as if total deal value would easily match 2015’s unprecedented levels.
But, much like Gatsby’s futile attempts to resurrect history, the medtech industry fell far short in recapturing its own past magic, as the blockbuster deals that defined the 2014-2015 gilded era dried up in the second half of 2017.
Nevertheless, M&A activity accelerated considerably this year in the orthopedic space as Globus Medical, NuVasive Inc., and Stryker Corp. arbitrated acquisitions that strengthened and/or boosted their scale in increasingly competitive therapeutic areas. Globus Medical’s midsummer deal for Swiss robotics developer KB Medical, for instance, is expected to help shape future enhancements to the larger firm’s ExcelsiusGPS robotic guidance and navigation system. KB Medical’s AQrate robotic system is used during lumbar fusion procedures to plan operations and guide instrument positioning for pedicle screw placement during the minimally invasive approach. The company received a CE mark for the AQrate system last year after completing a clinical trial treating more than 20 patients.
“The addition of KB Medical will enable Globus Medical to accelerate, enhance and expand our product portfolio in imaging, navigation and robotics,” president of Emerging Technologies Dave Demski said. “KB Medical’s team of technology development professionals, its strong IP portfolio and shared philosophy for robotic solutions in medicine strengthens Globus Medical’s position in this strategic area.”
Likewise, NuVasive’s purchase of Vertera Spine gives the company sole bragging rights to both polyetheretherketone (PEEK) and titanium porous interbody devices. Vertera’s biomaterial (Scoria) is grown from solid PEEK material, creating a seamless surface-to-solid material interface that is reportedly more durable than metal coatings and twice as strong as trabecular bone under shear loading.
Besides enhancing its PEEK technology offering, the Vertera deal also improves NuVasive’s chances of achieving its $1 billion revenue goal this year. The company moved well within range of its target with last year’s purchases of Ellipse Technologies, Biotronic NeuroNetwork, and LessRay software.
Better scale prompted other deals as well this year, enabling Marietta, Ga.-based Amendia to bolster its pedicle screw system offering and cervical product lines through Spinal Elements; Integra LifeSciences to expand its footprint in advanced wound care through Derma Sciences ($204 million); DePuy Synthes to accelerate spine market growth through the acquisition of Interventional Spine’s expandable cage technology; ConMed to strengthen its arthroscopy portfolio through the purchase of MedShape’s ExoShape Anterior Cruciate Ligament fixation system; and Stryker to fortify its advanced fluorescence imaging technology and minimally invasive surgical solutions for spine trauma via NOVADAQ Technologies Inc. ($701 million) and VEXIM ($216 million). Moreover, K2M Group Holdings Inc. padded its cervical spine showing by purchasing the PALO ALTO Cervical Static Corpectomy Cage System from Cardinal Spine, and enhanced its expandable interbody and 3D-printed product lineups through a patent portfolio agreement.
“...medtechs recognize that acquisitions that build end-to-end capabilities in a particular therapeutic area or expand the company’s geographic or technological reach are one of the fastest paths to growth,” EY analysts state in the firm’s “Pulse of the Industry 2017“ report.
But they’re not the only path to growth. Diversification inspired numerous transactions this year, pairing Avalign Technologies with Millennium Surgical and Thortex; Smith and Nephew plc with Rotation Medical Inc.; and DePuy Synthes with both Sentio LLC and Tissue Regeneration Systems. The latter deal gives DePuy the ability to develop personalized healthcare solutions in Trauma that potentially can help improve both patient satisfaction and clinical outcomes—key bellwethers in the migration toward value-based care.
Divestitures and spin-outs led to a few pairings this year too, most notably Johnson & Johnson and Integra LifeSciences. New Brunswick, N.J.-based J&J agreed in February to sell its Codman Neurosurgery business (sans the neurovascular and drug delivery divisions) for $1.05 billion to Integra LifeSciences, which has recently been restructuring its portfolio to build strong platforms in core areas like orthopedics and tissue technologies. J&J, meanwhile, has executed a similar strategy in its Medical Devices business, discarding low-performing segments to focus on higher growth markets such as orthopedics, surgery, and vision care.
“Divestitures and spin-outs allow medtechs to capture additional value by improving capital efficiency, reducing operational complexity, and reallocating capital to higher-growth businesses as the industry invests more R&D dollars in the development of innovative products that demonstrate value in an era of price pressures,” the EY report states. “Indeed...Johnson & Johnson’s divestiture of Codman Neurosurgery to Integra LifeSciences suggests capital efficiency and portfolio optimization will be an ongoing trend in 2018.”
Time will tell.
Read more: http://bit.ly/yir1708
Legal Troubles for DePuy
In the videos, they’re all smiling. Each and every one. There’s Rebecca, beaming broadly while walking hand-in-hand with two grandchildren; Roger, chuckling softly to his wife of 50 years; and Laura, laughing boldly and heartily during a trike motorcycle outing.
There’s also Bob, contentedly playing handball; Pam, cheerfully fixing a clothing store display; and Lani, grinning ever so slightly on horseback.
Absent from this sanguine sextet is Rocky Thompson of Pitkin, La., an unincorporated community of merely 576 residents. Thompson is not smiling. Nor is he happy.
Thompson could have been as happy as his counterparts: He replaced his ailing left knee with DePuy Synthes’ Attune Knee System, just like the others. But his recovery and post-replacement life has been markedly different than the merry band of Attune patient ambassadors featured on DePuy Synthes’ website.
Thompson blames the Attune system for his poor quality of life, claiming in a federal lawsuit that the product is defective and “unreasonably dangerous.” The suit, filed Sept. 26 in Louisiana district court, is reportedly the first to name Attune.
In the 43-page complaint, Thompson indicates he underwent knee replacement surgery in January 2015, swapping his natural joint for an Attune system comprised of a fixed tibial insert and fixed tibial baseplate. After the procedure, however, Thompson began experiencing “severe and persistent pain, discomfort, instability and difficulty ambulating” due to aseptic loosening and a tibial baseplate defect, according to the lawsuit. He underwent revision surgery in July this year—18 months after receiving the Attune joint.
Thompson’s lawsuit attributes the Attune’s defect to mechanical loosening, which is typically caused by a weakened or failing bond between the tibial baseplate and implant-cement surface. When such loosening occurs, the artificial knee can detach from existing bone, resulting in failure.
When it debuted in 2014, the Attune knee was considered an “advancement” in knee replacement options due to its S-curve design, gradual radius reduction, and a tibial base that optimizes kinematics while reducing wear. The technology was based on six years of research and inspired by overall high rates of patient dissatisfaction with artificial knees (up to 20 percent, industry-wide). In releasing Attune, DePuy promised patients greater functional benefits and a better range of motion compared to other implants.
Thompson, however, believes the product did more harm than good. “...in reality, the Attune device did not deliver on these promises, resulting in significantly higher failure rates than previous DePuy knee counterparts due to the debonding of the tibial baseplate,” his lawsuit states. “As a result, thousands of knee replacement patients implanted with Attune devices have had more expensive, more dangerous and less effective Total Knee Replacement surgeries, and many have required or will require expensive and dangerous knee revision surgery to remove and replace the defective Attune device.”
Although DePuy hasn’t responded directly to Thompson’s lawsuit, the company has released volumes of Attune study data confirming low revision rates and high implant survivorship. Statistics from the National Joint Registry for England, Wales, Northern Ireland and the Isle of Man (NJR), for example, show an estimated cumulative revision rate of 1.3 percent at four years (98.7 percent implant survivorship) for 10,605 Attune knees, which compares favorably to the 1.9 percent cumulative revision rate (98.1 percent implant survivorship) for the overall class of knee replacements.
The numbers are even better Down Under: The Attune Cruciate Retaining Knee boasts a 2.1 percent cumulative revision rate at three years, which is consistent with the class, while the Posterior Stabilized Knee exhibited a 1.1 percent rate at three years, which compares favorably to the class, according to data from 8,3841 Attune knees tracked by the Australian Orthopaedic Association National Joint Replacement Registry. Attune also scored well in New Zealand, claiming an overall 0.66 revision rate per 100 component years, with 33 revisions (of 3,795 implants), data from an 18-year joint registry report show.
The numbers are solid in Michigan as well, with the Attune knee showing a 2.42 percent revision rate after three years based on 4,870 implants from 2012 to 2016, the Michigan Arthroplasty Registry Collaborative Quality Initiative (MARQI) concluded.
Furthermore, a radiostereometric (RSA) analysis conducted by the Canadian RSA Network found the Attune tibial base to be stable, moving only 0.17 mm between one and two years. The results are consistent with implants that have acceptable revision rates due to aseptic loosening. A second, randomized-controlled RSA study conducted in the Netherlands compared Attune implant migration to patients implanted with SIGMA knees and reported no difference in implant migration.
“Attune has been in clinical use for over six years now, and we have over half a million cases [of it used] around the world,” Rajit Kamal, global knee platform leader for DePuy Synthes, told Orthopedic Design & Technology. “There’s a solid body of evidence that very clearly shows that Attune is delivering on the intended objectives we had. It is showing better patient-reported outcomes, patient satisfaction, and implant survivorship in-line with the class.”
Maybe so, but a growing body of evidence suggests otherwise. Thompson’s lawsuit cites 1,400 reports of Attune knee failures from the U.S. Food and Drug Administration’s (FDA) MAUDE (Manufacturer and User Facility Device Experience) database, with roughly 633 of those failures resulting in revision surgeries. The suit, however, does not specify a reason for either the failures or the revisions.
Similarly, an April 2017 study published in the Journal of Knee Surgery shows a “high” rate of Attune tibial loosening at the implant-cement interface. Researchers identified 15 cases from three hospitals and referenced 21 additional reports of tibial loosening in the MAUDE database within a two-month period as well as “numerous other tibial failures” also reported without the mechanism for failure specified.
DePuy, of course, questioned the study’s accuracy, noting the absence of a rate calculation and radiographic technique or positioning. The company also took issue with the researchers’ use of MAUDE database statistics, claiming it conflicts with the intent of the database and FDA guidance. “The FDA’s guidance is that, ‘MDR data alone cannot be used to establish rates of events, evaluate a change in event rates over time or compare event rates between devices. The number of reports cannot be interpreted or used in isolation to reach conclusions about the existence, severity, or frequency of problems associated with devices,’ “DePuy said in a formal response to the Journal of Knee Surgery study.
The company’s defense resonated with Cleveland Clinic doctor Michael A. Mont, who criticized the study for its lack of a denominator. “When one looks at a case series, we really do not know the denominator, and this certainly could have been a series of cases out of more than 100,000 done, which would make this a rare event,” Mont wrote in a Journal opinion piece. “Therefore, simply publishing cases is not evidence-based medicine, as we do not know how many of these were implanted. For example, ceramic fractures occur very infrequently, maybe one in 10,000; and yet, a small series could easily be published. The key is to know the denominator. Although this report may raise some interest, I would like to think that it is only an isolated group of failures that can occur with any device in the field from any manufacturer.”
Perhaps, but Thompson disagrees. He contends the Attune knee’s tibial base plate is defective, and points to DePuy’s newest iteration, the Attune S+ as proof. “Defendants’ research, design, marketing, and placement of the Attune S+ with new design features on the market aimed at increasing fixation is an admission that the original Attune tibial base plate was defective in its composition and/or construction,” he argues in his lawsuit.
DePuy, though, dismisses such allegations, asserting the Attune S+ is designed to further improve the industry-wide issue of aseptic loosening rates. The S+ technology, in development for the last two years, combines macrolock features and a microblast finish to inhibit lipid infiltration on the tibial base surface and increase pull-off strength under common simulated intra-operative surgical conditions. The product’s rougher surface is intended to increase cement-implant interdigitation and reduce the amount of fluids that might bleed into the cement-implant interface. In addition, four cement pockets with 45-degree undercuts and macro geometry aim to improve mechanical fixation with cured cement.
“Based on the literature out there, benchtop testing, and cadaveric studies, we found that the biggest variable that causes a reduction of the strength of fixation between the inner part [of the implant] and the cement is the introduction of lipids or marrow between those two components (cement and implant),” Liam Rowley, worldwide product director, Knees, for DePuy Synthes, noted to ODT. “We found that a combination of a macrolock plus an optimized microblast—the combination of those two together—makes the product much more resistant to lipids infiltration. So far, the Attune S+ has outperformed everything we’ve measured against in terms of benchtop testing. We’re really excited about it, because we think we’ve got a product that will really raise the bar for the whole industry in terms of tibial fixation.”
Read more: http://bit.ly/yir1711