Mike Goodson, Associate Director, Regulatory Affairs, MCRA LLC09.17.19
As of 2017, the European medical device market was estimated at $126 billion, the second largest worldwide, comprising 27 percent of the global market. The European market trails only the United States, which accounts for roughly 43 percent of the global medical device market, according to a MedTech Europe publication.1 When the new Medical Device Regulations (MDR 2017/745) takes effect on May 26, 2020, increased requirements and higher levels of scrutiny related to almost all aspects of the medical device product lifecycle are expected. In particular, the process related to demonstrating compliance with the applicable clinical requirements has intensified under the upcoming MDR, putting companies at risk of being unable to recertify or introduce their devices into Europe under the new regulations.
Manufacturers were granted a three-year transition period from the date the MDR was officially published. Medical devices approved in Europe under the current legislation (the Directives) can still be placed on the market until their certificate expires, or no later than May 26, 2024. This is valid only if the provisions laid out in Article 120 of the MDR are applicable and followed. As such, manufacturers are racing to recertify and/or extend their certificates’ validity date under the current Directives to give themselves more time to recertify under the MDR’s full requirements. To avoid potential market disruption in Europe, manufacturers should be assessing the clinical data captured for their devices now under the Directives and start planning post-market clinical follow-up (PMCF) activities not only to close gaps in their current clinical data, but to also meet the MDR’s proactive post-market surveillance (PMS)/PMCF requirements.
This column will identify common deficiencies found when assessing the clinical evaluation of a device and how a PMCF can address these deficiencies, close data gaps, obtain long-term data, and meet MDR requirements.
The requirement for a medical device to have sufficient clinical data as part of the conformity assessment in Europe is not new, nor is the requirement to have clinical data confirming the medium or long-term safety and performance of the device throughout its expected lifetime. In 2012, the existing PMCF guidance document was revised, emphasizing the need for long-term clinical data and developing a risk-based PMS plan as well as highlighting the importance of having a PMCF plan that provides the detailed documentation required for post-market clinical follow-up activities.
In June 2016, the clinical evaluation requirements for medical devices in Europe took a dramatic shift with the release of a revised clinical evaluation guidance document. This guidance emphasized the need for clinical data on the manufacturers’ actual device as seen through detailed and prescriptive requirements around demonstrating clinical compliance.
The MDR, much like the PMCF guidance and the clinical evaluation guidance, places a greater importance on the collection of clinical data on the manufacturer’s own device through a proactive and systematic approach over the lifetime of the device, especially in post-market surveillance.
Clinical Evaluation Gap Analysis
To determine what the scope of a PMCF may look like, manufacturers should perform a thorough gap analysis on all available clinical data captured in their device’s clinical evaluation report (CER). Gap analyses should be performed in line with the new requirements. This is true even for manufacturers with valid certificates under one of the Directives who intend to continue to place devices in Europe. When performing a gap analysis, manufacturers have several items to consider when determining whether the clinical data on their device(s) are sufficient. These considerations include, but are not limited to:
PMCF
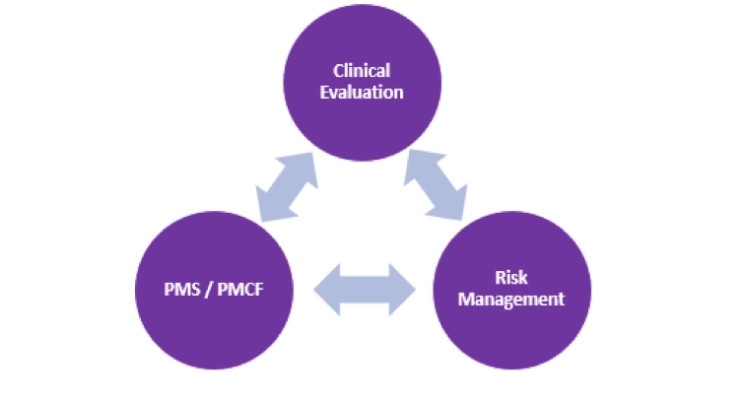
Devices with valid certificates under the applicable Directives can be placed on the European market after the MDR goes into effect; however, certain requirements of the MDR apply in place of the corresponding Directives requirements. The post-market surveillance (PMS)/PMCF requirements demand manufacturers be in compliance when the MDR goes into effect, irrespective of the device’s risk class. PMCF is a substantial part of PMS under the MDR. The PMCF and a documented PMCF plan are required for all devices. The PMCF is a continuous process that updates the clinical evaluation and ensures continued acceptability of identified risks. Simply put, the PMCF needs to be aligned with the clinical evaluation and risk documentation so data from all documents feed into each other. The PMCF can then be adjusted based on the output of the clinical evaluation and risk documentation.
The PMCF Plan
The MDR provides the following requirements for the PMCF plan:
“The PMCF plan shall include at least:
Circumstances Justifying a PMCF
PMCF may be required in the following circumstances:
When a manufacturer already has sufficient clinical data related to the long-term performance and safety of the device or where other appropriate post-market surveillance activities would provide sufficient data to address, a PMCF study may not be required. A decision by the manufacturer to not perform a PMCF must be duly justified and documented in the PMS documentation.
PMCF Study Design
The PMCF is designed to answer specific questions about clinical safety or device performance (i.e. residual risks) when used in accordance with its approved labeling. PMCF activities are required to be conducted according to applicable laws and regulations, and should involve an appropriate methodology and follow appropriate guidance and standards. The activities will vary based on the objective(s) defined in the PMCF plan. By definition, the PMCF collects clinical data proactively, and methodologies to proactively collect clinical data include extended patient follow-up in premarket investigations; a new clinical investigation; review of data derived from a device registry; review of relevant retrospective data from patients previously exposed to the device; and planned customer surveys.
The PMCF does not have to be a large randomized trial with a control group and strict inclusion/exclusion criteria, but clinical data from a randomized controlled trial should result in the highest data appraisal scores. Manufacturers should select a PMCF design to ensure the activities are appropriate to answer the PMCF plan objectives. For example, if long-term clinical data are needed based on the results of a gap analysis, a PMCF in the form of customer surveys/questionnaires might not be the best study design to capture the required data.
Conclusion
A detailed and thorough gap analysis of a device’s clinical data will help determine the PMCF scope. To avoid budget busting studies, a clearly defined PMCF objective and plan should be established and strictly followed. Establishing an MDR-compliant PMCF should begin now for devices certified under the Directives to capture data that will be accepted when recertifying under the MDR.
PMCF activities can provide manufacturers with important data related to device safety such as confirming the acceptably of current risks and reducing residual risks to acceptable levels, which greatly benefits patients. Finally, long-term clinical data will provide manufacturers with robust clinical evidence which can help increase market share as well as payer reimbursement.
Reference
Mike Goodson is an associate director, Regulatory Affairs, at MCRA LLC, focusing on developing regulatory strategies and submissions to European Notified Bodies and the FDA. He has knowledge of regulatory pathways with experience developing EU Technical Files including Clinical Evaluation Reports, and PMAs, IDEs, 510(k)s, and de novo submissions, with a focus in orthopedic devices.
Manufacturers were granted a three-year transition period from the date the MDR was officially published. Medical devices approved in Europe under the current legislation (the Directives) can still be placed on the market until their certificate expires, or no later than May 26, 2024. This is valid only if the provisions laid out in Article 120 of the MDR are applicable and followed. As such, manufacturers are racing to recertify and/or extend their certificates’ validity date under the current Directives to give themselves more time to recertify under the MDR’s full requirements. To avoid potential market disruption in Europe, manufacturers should be assessing the clinical data captured for their devices now under the Directives and start planning post-market clinical follow-up (PMCF) activities not only to close gaps in their current clinical data, but to also meet the MDR’s proactive post-market surveillance (PMS)/PMCF requirements.
This column will identify common deficiencies found when assessing the clinical evaluation of a device and how a PMCF can address these deficiencies, close data gaps, obtain long-term data, and meet MDR requirements.
The requirement for a medical device to have sufficient clinical data as part of the conformity assessment in Europe is not new, nor is the requirement to have clinical data confirming the medium or long-term safety and performance of the device throughout its expected lifetime. In 2012, the existing PMCF guidance document was revised, emphasizing the need for long-term clinical data and developing a risk-based PMS plan as well as highlighting the importance of having a PMCF plan that provides the detailed documentation required for post-market clinical follow-up activities.
In June 2016, the clinical evaluation requirements for medical devices in Europe took a dramatic shift with the release of a revised clinical evaluation guidance document. This guidance emphasized the need for clinical data on the manufacturers’ actual device as seen through detailed and prescriptive requirements around demonstrating clinical compliance.
The MDR, much like the PMCF guidance and the clinical evaluation guidance, places a greater importance on the collection of clinical data on the manufacturer’s own device through a proactive and systematic approach over the lifetime of the device, especially in post-market surveillance.
Clinical Evaluation Gap Analysis
To determine what the scope of a PMCF may look like, manufacturers should perform a thorough gap analysis on all available clinical data captured in their device’s clinical evaluation report (CER). Gap analyses should be performed in line with the new requirements. This is true even for manufacturers with valid certificates under one of the Directives who intend to continue to place devices in Europe. When performing a gap analysis, manufacturers have several items to consider when determining whether the clinical data on their device(s) are sufficient. These considerations include, but are not limited to:
- Does the clinical data cover the stated device lifetime?
- Are there sufficient data that support the full intended use(s) of the device?
- Does the clinical data support device claims/benefits?
- Do the data appraisal results satisfy requirements?
- Are the data from studies conducted in compliance with current quality and ethical standards such as, Good Clinical Practices (GCP), EN ISO 14155, and the Declaration of Helsinki?
PMCF
Devices with valid certificates under the applicable Directives can be placed on the European market after the MDR goes into effect; however, certain requirements of the MDR apply in place of the corresponding Directives requirements. The post-market surveillance (PMS)/PMCF requirements demand manufacturers be in compliance when the MDR goes into effect, irrespective of the device’s risk class. PMCF is a substantial part of PMS under the MDR. The PMCF and a documented PMCF plan are required for all devices. The PMCF is a continuous process that updates the clinical evaluation and ensures continued acceptability of identified risks. Simply put, the PMCF needs to be aligned with the clinical evaluation and risk documentation so data from all documents feed into each other. The PMCF can then be adjusted based on the output of the clinical evaluation and risk documentation.
The PMCF Plan
The MDR provides the following requirements for the PMCF plan:
“The PMCF plan shall include at least:
- The general methods and procedures of the PMCF to be applied, such as gathering of clinical experience gained, feedback from users, screening of scientific literature and of other sources of clinical data;
- The specific methods and procedures of PMCF to be applied, such as evaluation of suitable registers or PMCF studies;
- A rationale for the appropriateness of the methods and procedures
- A reference to the relevant parts of the clinical evaluation report and to the risk management
- The specific objectives to be addressed by the PMCF;
- An evaluation of the clinical data relating to equivalent or similar devices;
- Reference to any relevant harmonized standards when used by the manufacturer, and relevant guidance on PMCF; and
- A detailed and adequately justified time schedule for PMCF activities (e.g. analysis of PMCF data and reporting) to be undertaken by the manufacturer.”
Circumstances Justifying a PMCF
PMCF may be required in the following circumstances:
- Innovation, e.g., where device design, the materials, substances, principles of operation, technology or the medical indications are novel;
- Significant changes to the product or to its intended use for which premarket clinical evaluation and re-certification has been completed;
- High product related risk based on design, materials, components, invasiveness, clinical procedures;
- High risk anatomical locations;
- High risk target populations e.g. pediatrics, elderly;
- Severity of disease/treatment challenges;
- Questions of ability to generalize clinical investigation results;
- Unanswered questions of long-term safety and performance;
- Results from any previous clinical investigation, including adverse events or from post-market surveillance activities;
- Verification of safety and device performance when exposed to a larger and more varied clinical user population;
- Emergence of new information on safety or performance;
- Where CE marking was based on equivalence.
When a manufacturer already has sufficient clinical data related to the long-term performance and safety of the device or where other appropriate post-market surveillance activities would provide sufficient data to address, a PMCF study may not be required. A decision by the manufacturer to not perform a PMCF must be duly justified and documented in the PMS documentation.
PMCF Study Design
The PMCF is designed to answer specific questions about clinical safety or device performance (i.e. residual risks) when used in accordance with its approved labeling. PMCF activities are required to be conducted according to applicable laws and regulations, and should involve an appropriate methodology and follow appropriate guidance and standards. The activities will vary based on the objective(s) defined in the PMCF plan. By definition, the PMCF collects clinical data proactively, and methodologies to proactively collect clinical data include extended patient follow-up in premarket investigations; a new clinical investigation; review of data derived from a device registry; review of relevant retrospective data from patients previously exposed to the device; and planned customer surveys.
The PMCF does not have to be a large randomized trial with a control group and strict inclusion/exclusion criteria, but clinical data from a randomized controlled trial should result in the highest data appraisal scores. Manufacturers should select a PMCF design to ensure the activities are appropriate to answer the PMCF plan objectives. For example, if long-term clinical data are needed based on the results of a gap analysis, a PMCF in the form of customer surveys/questionnaires might not be the best study design to capture the required data.
Conclusion
A detailed and thorough gap analysis of a device’s clinical data will help determine the PMCF scope. To avoid budget busting studies, a clearly defined PMCF objective and plan should be established and strictly followed. Establishing an MDR-compliant PMCF should begin now for devices certified under the Directives to capture data that will be accepted when recertifying under the MDR.
PMCF activities can provide manufacturers with important data related to device safety such as confirming the acceptably of current risks and reducing residual risks to acceptable levels, which greatly benefits patients. Finally, long-term clinical data will provide manufacturers with robust clinical evidence which can help increase market share as well as payer reimbursement.
Reference
Mike Goodson is an associate director, Regulatory Affairs, at MCRA LLC, focusing on developing regulatory strategies and submissions to European Notified Bodies and the FDA. He has knowledge of regulatory pathways with experience developing EU Technical Files including Clinical Evaluation Reports, and PMAs, IDEs, 510(k)s, and de novo submissions, with a focus in orthopedic devices.