Dan Goldstein, Associate Director, Quality Assurance, MCRA LLC12.02.19
When any new regulation or standard takes effect, the companies that quickly get on board gain a competitive advantage over those who take longer. Waiting to comply at the last minute often costs extra time and money, and the self-sorting of companies between early and late adopters is sometimes visible to customers—not to mention potential investors and acquirers.
However, with the new European Union Medical Device Regulation (EU MDR), there are additional benefits rooted in the contents of the regulation. This column examines three new elements of the EU MDR that medical device manufacturers can leverage. Each case will show how regulatory, quality, and clinical staff can help companies make competitive use of the new regulation.
The Summary of Safety and Clinical Performance
Under the EU MDR, the European Database on Medical Devices (EUDAMED) will make data publicly available that, until now, was only reported confidentially by manufacturers to their Notified Bodies. This includes the new Summary of Safety and Clinical Performance (SSCP), which will be required for every implantable and class III device marketed in the EU. The following table lists some selected SSCP data inputs, and how they can be leveraged by a manufacturer seeking a new CE mark.
In addition to the SSCPs, EUDAMED will also make publicly available some vigilance data from competing devices.3 This previously confidential data includes serious reportable incidents and Field Safety Corrective Actions (FSCAs). As with the SSCP data, this vigilance data can be used to justify risk calculations and guide the new device’s design.
Economic Operators and Single Registration Numbers
“Economic Operator” is another new concept under the EU MDR. Economic Operators include all device manufacturers and their European distributors, regardless of device classification. For manufacturers located outside the EU, Economic Operators also include importers and Authorized Representatives.
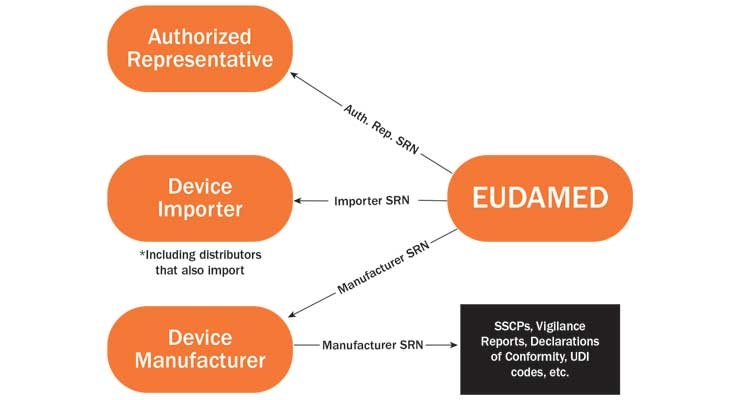
Here, too, EUDAMED will offer far more information than has been publicly available until now in the EU. With the exception of distributors, Economic Operators will be required to register with Single Registration Numbers (SRNs) assigned by EUDAMED so each device can be linked with its Economic Operators. All of this information will be readily searchable by the public.
Just as SSCPs are set to reveal previously hidden information about the devices themselves, the SRNs of devices and Economic Operators will reveal previously hidden information about imported devices’ supply chains. Similar information is currently available about commercial distribution of imported devices in the United States, through registration information on the U.S. Food and Drug Administration’s (FDA) website. One significant difference from the United States is the country-specific location of the publicly registered importer into the EU: The new MDR applies across the EU, but each country is still a unique market, and information about competitors’ choices for first countries of distribution could inform marketing strategies for new devices.
Expert Panel to Support Clinical Trial Goals
The EU MDR features several innovations designed to increase medical, scientific, and technical scrutiny of new devices. Among these innovations are the new Expert Panels, which will be created for all Class III and certain Class IIb devices. The panels are required to include advisors from across the EU with “up-to-date clinical, scientific or technical expertise.”4
In theory, the Expert Panels have only an advisory role, but the EU MDR makes it hard for a Notified Body to ignore an Expert Panel’s advice. For example:
Despite the concerns that Expert Panels pose for Notified Bodies, manufacturers should consider Expert Panels as an opportunity for competitive advantage. The EU MDR encourages a manufacturer to consult expert panels prior to developing clinical evaluations and investigations, in order to get insight “on its clinical development strategy and on proposals for clinical investigations.”6 Under the old MDD, a manufacturer could hold similar discussions with Notified Bodies, but only with careful boundaries around certain consultancy services.
Under the EU MDR, consulting with an Expert Panel allows a manufacturer to glean knowledge and guidance from the very experts the Notified Body will find it hard to disagree with. Not only that, but Expert Panels will have extensive knowledge of past clinical investigations on competing devices, which could help the manufacturer focus their new clinical work for the greatest effect at the least expense.
Under the EU MDR, EUDAMED will offer significantly more information than has been publicly available until now in Europe.
Conclusion
These scenarios are only a partial list of EU MDR elements that can be used for competitive advantage. For example, with the pool of available Notified Bodies expected to shrink dramatically in the short term, a manufacturer that moves aggressively to recertify devices under the EU MDR could get ahead of the expected logjam as the final MDD certificates begin to expire. Also, the Medical Device Single Audit Program (MDSAP) could also have greater value in a market where the Notified Bodies are harder pressed to perform quality management system (QMS) assessments in time. Finally, the suddenly public EUDAMED is likely to reveal further competitive advantages as new guidances are written to better define the data submission requirements.
Until now, the commercially actionable EU data for manufacturers seeking a new CE mark was limited—generally less than is available from FDA for parallel purposes in the United States. Under the EU MDR, future marketing in the EU could be based on significantly more data than is available for the United States.
Of course, any valuable new data can require hard work to obtain and process. A recent ODT article described the EU MDR and its opaque, accelerated rollout as a “disaster” for the medical device industry. Still, while it’s true that the MDR’s greatly expanded regulatory framework has created new challenges for getting to market, these new challenges also offer new opportunities.
References
Dan Goldstein is the associate director for Quality Assurance at MCRA LLC, focusing primarily on quality system and manufacturing requirements, in order to keep devices and manufacturers in compliance with U.S. and international regulations and standards. A graduate of the University of Maryland University College, Dan has worked in medical device quality assurance since 2002 and for MCRA since 2016. Dan provides MCRA clients with FDA QSR and EU MDR gap assessments, mock FDA inspections, internal and supplier audits, manufacturing transfers, due diligence reporting, Form 483 remediations, Medical Device Reporting, Unique Device Identification, recall support, Design History Files, and Technical Documentation. MCRA LLC has experience providing international regulatory services. MCRA’s consultants provide the knowledge and experience to support medical device companies looking to gain access to established and emerging markets. MCRA can assist with Notified Body identification, quality system and manufacturing compliance (e.g. ISO 13485:2016), MDSAP preparation and implementation, internal audits, supplier audits, and clinical investigation services, including pre-study activities and study execution.
However, with the new European Union Medical Device Regulation (EU MDR), there are additional benefits rooted in the contents of the regulation. This column examines three new elements of the EU MDR that medical device manufacturers can leverage. Each case will show how regulatory, quality, and clinical staff can help companies make competitive use of the new regulation.
The Summary of Safety and Clinical Performance
Under the EU MDR, the European Database on Medical Devices (EUDAMED) will make data publicly available that, until now, was only reported confidentially by manufacturers to their Notified Bodies. This includes the new Summary of Safety and Clinical Performance (SSCP), which will be required for every implantable and class III device marketed in the EU. The following table lists some selected SSCP data inputs, and how they can be leveraged by a manufacturer seeking a new CE mark.
| Selected SSCP Data Input1 | How to Leverage for a New Device |
|---|---|
| Intended purpose, indications, and contraindications | Identify similar devices for potential equivalence to the new device.2 |
| Initial CE marking date | Identify the latest approved technologies by sorting similar devices by market entry date. |
| Residual risks, undesirable effects, and warnings, including quantifiable probability of harm | Use competing devices’ data to streamline and justify risk calculations for the new device; design the new device competitively to reduce occurrence of identified hazards. |
| Summary of Clinical Evaluation Report (CER) and Post-Market Clinical Follow-up (PMCF) | Use competing devices’ CER and PMCF summaries to determine and justify extent of the new device’s pre- and post-market clinical studies. |
| Harmonized standards and Common Specifications (CSs) | Use competing devices’ citations of standards and CSs to determine and justify “state-of-the-art” requirements for the new device’s General Safety and Performance Requirements (GSPR). |
In addition to the SSCPs, EUDAMED will also make publicly available some vigilance data from competing devices.3 This previously confidential data includes serious reportable incidents and Field Safety Corrective Actions (FSCAs). As with the SSCP data, this vigilance data can be used to justify risk calculations and guide the new device’s design.
Economic Operators and Single Registration Numbers
“Economic Operator” is another new concept under the EU MDR. Economic Operators include all device manufacturers and their European distributors, regardless of device classification. For manufacturers located outside the EU, Economic Operators also include importers and Authorized Representatives.
Here, too, EUDAMED will offer far more information than has been publicly available until now in the EU. With the exception of distributors, Economic Operators will be required to register with Single Registration Numbers (SRNs) assigned by EUDAMED so each device can be linked with its Economic Operators. All of this information will be readily searchable by the public.
Just as SSCPs are set to reveal previously hidden information about the devices themselves, the SRNs of devices and Economic Operators will reveal previously hidden information about imported devices’ supply chains. Similar information is currently available about commercial distribution of imported devices in the United States, through registration information on the U.S. Food and Drug Administration’s (FDA) website. One significant difference from the United States is the country-specific location of the publicly registered importer into the EU: The new MDR applies across the EU, but each country is still a unique market, and information about competitors’ choices for first countries of distribution could inform marketing strategies for new devices.
Expert Panel to Support Clinical Trial Goals
The EU MDR features several innovations designed to increase medical, scientific, and technical scrutiny of new devices. Among these innovations are the new Expert Panels, which will be created for all Class III and certain Class IIb devices. The panels are required to include advisors from across the EU with “up-to-date clinical, scientific or technical expertise.”4
In theory, the Expert Panels have only an advisory role, but the EU MDR makes it hard for a Notified Body to ignore an Expert Panel’s advice. For example:
- If a Notified Body certifies a device against the Expert Panel’s advice, the Notified Body must provide a justification in writing to the Competent Authorities.
- EUDAMED maintains a public record of each expert panel’s opinions, along with every written justification for a Notified Body diverging from that opinion.
- The European Parliament receives an annual report of every case where a Notified Body chooses not to follow the advice of an Expert Panel.5
Despite the concerns that Expert Panels pose for Notified Bodies, manufacturers should consider Expert Panels as an opportunity for competitive advantage. The EU MDR encourages a manufacturer to consult expert panels prior to developing clinical evaluations and investigations, in order to get insight “on its clinical development strategy and on proposals for clinical investigations.”6 Under the old MDD, a manufacturer could hold similar discussions with Notified Bodies, but only with careful boundaries around certain consultancy services.
Under the EU MDR, consulting with an Expert Panel allows a manufacturer to glean knowledge and guidance from the very experts the Notified Body will find it hard to disagree with. Not only that, but Expert Panels will have extensive knowledge of past clinical investigations on competing devices, which could help the manufacturer focus their new clinical work for the greatest effect at the least expense.
Under the EU MDR, EUDAMED will offer significantly more information than has been publicly available until now in Europe.
Conclusion
These scenarios are only a partial list of EU MDR elements that can be used for competitive advantage. For example, with the pool of available Notified Bodies expected to shrink dramatically in the short term, a manufacturer that moves aggressively to recertify devices under the EU MDR could get ahead of the expected logjam as the final MDD certificates begin to expire. Also, the Medical Device Single Audit Program (MDSAP) could also have greater value in a market where the Notified Bodies are harder pressed to perform quality management system (QMS) assessments in time. Finally, the suddenly public EUDAMED is likely to reveal further competitive advantages as new guidances are written to better define the data submission requirements.
Until now, the commercially actionable EU data for manufacturers seeking a new CE mark was limited—generally less than is available from FDA for parallel purposes in the United States. Under the EU MDR, future marketing in the EU could be based on significantly more data than is available for the United States.
Of course, any valuable new data can require hard work to obtain and process. A recent ODT article described the EU MDR and its opaque, accelerated rollout as a “disaster” for the medical device industry. Still, while it’s true that the MDR’s greatly expanded regulatory framework has created new challenges for getting to market, these new challenges also offer new opportunities.
References
- MDCG 2019-9 Summary of Safety and Clinical Performance: A guide for manufacturers and notified bodies. EU Medical Device Coordination Group, August 2019.
- Unlike the old MDD, equivalence under the EU MDR requires the two manufacturers to have a contract that allows ongoing mutual access to technical documentation.
- EU MDR (2017/745) Article 92(3)
- EU MDR (2017/745) Article 106(3)
- EU MDR (2017/745) Article 54(4)
- EU MDR (2017/745) (57)
Dan Goldstein is the associate director for Quality Assurance at MCRA LLC, focusing primarily on quality system and manufacturing requirements, in order to keep devices and manufacturers in compliance with U.S. and international regulations and standards. A graduate of the University of Maryland University College, Dan has worked in medical device quality assurance since 2002 and for MCRA since 2016. Dan provides MCRA clients with FDA QSR and EU MDR gap assessments, mock FDA inspections, internal and supplier audits, manufacturing transfers, due diligence reporting, Form 483 remediations, Medical Device Reporting, Unique Device Identification, recall support, Design History Files, and Technical Documentation. MCRA LLC has experience providing international regulatory services. MCRA’s consultants provide the knowledge and experience to support medical device companies looking to gain access to established and emerging markets. MCRA can assist with Notified Body identification, quality system and manufacturing compliance (e.g. ISO 13485:2016), MDSAP preparation and implementation, internal audits, supplier audits, and clinical investigation services, including pre-study activities and study execution.