Dr. Sherry Parker and Dr. Kimberly Ehman, WuXi AppTec Medical Device Testing05.17.21
With its unique properties, nitinol is a popular material choice for medical devices but there are critical factors to keep in mind when considering potential risk and patient safety. More than 50 percent of nitinol is nickel, and nickel-ion release can lead to sensitization as well as systemic and local tissue effects. Manufacturers using at least one nitinol device component must consider recent U.S. Food and Drug Administration (FDA) guidance and protect end users from nickel-ion release.
The FDA's “Technical Considerations for Non-Clinical Assessment of Medical Devices Containing Nitinol” covers guidance in four primary areas: mechanical testing, corrosion, biocompatibility, and labeling. Identifying and resolving potential biocompatibility concerns when corrosion is a possibility can be challenging, especially with devices that were not previously required to meet this regulatory expectation. Standard chemical characterization testing cannot predict the potential toxicological effects of nickel ion release over time, which leaves a gap to fill during safety evaluation.
The Need for Nickel Ion Release Evaluations
The way the medical device industry evaluates products from a patient safety standpoint is constantly evolving. Nitinol differs from other metals that manufacturers might use in medical devices like stainless steel or titanium. Specifically, nitinol has shape memory qualities and flexibility that are difficult to replace with alternative materials.
The FDA’s guidance states, "Given the complex thermomechanical behavior of nitinol, there are unique considerations when assessing the safety and performance of nitinol devices."
Required methods that would traditionally identify material risks such as chemical characterization and toxicological risk assessments (TRAs) cannot fully capture its nickel release concerns. Therein, the need for alternative approaches remains. When a device has the potential for corrosion, and particularly when it is an implantable device, regulators are looking to see expanded testing to evaluate the risks of adverse health effects.
Nickel ion release is the primary concern with the presence of nitinol. Surface processing can have a significant impact on corrosion and nickel ion release. If a device does not have an established surface finish, manufacturers may need to perform an in-vitro nickel ion release study. Understanding the potential for nickel ion release may help the manufacturer optimize the manufacturing process if it decides to take a different approach with the surface finish of nitinol.
Key Call Outs in FDA Guidance Updates
Nitinol has long been a red flag for regulators evaluating cardiovascular devices, and with the increased interest in using minimally invasive procedures, nitinol applications have expanded.
Transcatheter heart valves, orthodontic devices, stents, and orthopedic fracture fixation (e.g., plates, screws, staples) are all examples of medical devices the FDA will be watching for evidence that nickel release is not a concern.
Manufacturers should evaluate any device containing nitinol that has either prolonged contact or long-term patient exposure under the latest guidance. Limited contact devices are considered at lower risk for nickel ion release and may not require the same testing. But any limited contact devices with an electrically active component, dissimilar metal couple or degradable metal/polymer components may also need to address nitinol risks under this guidance. In short, the test requirements for nickel ion release now include many more types of devices beyond just cardiovascular devices.
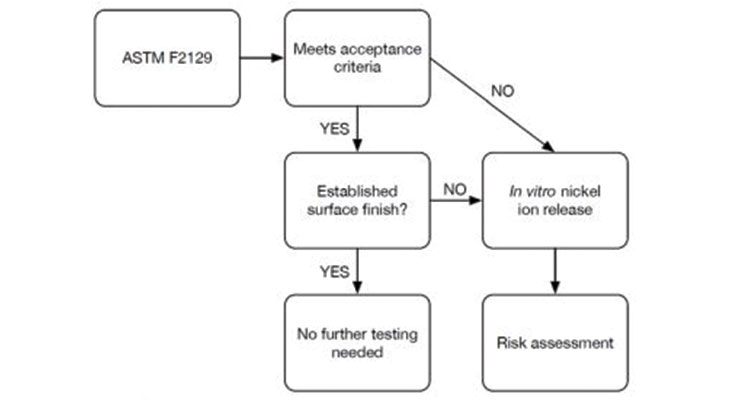
Manufacturers with nitinol-containing devices seeking advice on meeting regulatory expectations should consult the recent guidance. As shown by Figure 1, the addition of a corrosion testing flowchart acts as a valuable resource to understand expectations. This chart demonstrates where to look for corrosion testing acceptance criteria (ASTM F2129) and the steps to follow if testing does not meet criteria, or meets criteria but does not have an established surface finish. If additional testing is needed, manufacturers should use in-vitro nickel ion release methods to support the risk assessment.
Considerations for Prolonged and Long-Term Contacting Devices
Manufacturers can avoid costly delays by recognizing the pros and cons of each material choice before beginning safety testing. This foresight is crucial for prolonged or long-term contacting devices due to the heightened expectations. Even then, the slightest of changes to the manufacturing process can impact surface properties. These cases and any other corrosion possibility can affect a device's biocompatibility and the necessary testing requirements. If companies make manufacturing changes, consider additional testing and communicate goals with testing partners at the onset.
When laboratory testing partners have experience with nitinol and the testing needed, they can help avoid repeat studies and other potential delays. The investments in both biocompatibility and chemical characterization testing for implantable devices are substantial, and the last thing anyone wants is lack of communication to be the detrimental downfall.
The Limitations of Biocompatibility Testing Methods
Systemic toxicity is a concern for leachable metal ions, and while the FDA guidance document recommends an acceptable level for a TRA, this does not address sensitization concerns. The route and duration of exposure are key inputs to systemic toxicity; however, this endpoint can be addressed in a TRA because there is an established acceptable daily exposure.
Alternatively, sensitization is not easy to predict and functions in a manner much like an allergic reaction. The immune response leading to sensitization is variable between animal models and humans, and across the human population as a whole. Consequently, the use of different testing systems is not always predictive of the human response. Due to variability, there is no known lower limit on the amount of nickel that can elicit a sensitization response in some patients or be applied in the TRA.
The risk assessment of the chemical characterization and nickel ion release studies can address potential systemic toxicity to nickel. Still, regulators continue to view in-vivo models as the gold standard for addressing possible sensitization. Additionally, the guidance recommends potential risk mitigation through labeling, particularly for prolonged and long-term contacting devices.
Manufacturers who are still unsure how testing recommendations apply to their device can get feedback from regulators using a Q-sub process outlined in the guidance updates for direct consultation. The guidance also provides additional resources to help companies get started on this process.
Emerging Approaches to Protect Patient Safety
Protecting patient safety has taken new forms in recent years with a shift to more customized testing that addresses device-specific risks rather than a checklist approach. This guidance speaks to a clear theme of regulators looking into the potential material risks of each device. This trend is also evident in standards like ISO 10993-18:2020, which guides medical device biological safety testing through more detailed chemical characterization to support the evaluation of toxicological risks.
Additionally, a wide range of devices is now included under the standard, representing even more device categories and patient exposures than before. In particular, prolonged exposure and long-term contacting devices are the most affected by this guidance. For many companies, this might be a learning curve, but fortunately, they have resources including previous knowledge from the cardiovascular category and support from partners such as testing laboratories.
Before embarking on upcoming medical device safety testing, manufacturers should vet any potential impact from nitinol components on their test plan. Understanding the risks associated with each material choice is key, and customizing testing to fit this helps satisfy regulator questions.
Dr. Parker has more than 20 years of toxicology and medical device experience, and is an expert in biological evaluation of medical devices and combination products. She received her Ph.D. in molecular and cellular pharmacology from the University of Miami. In her current position as WuXi AppTec’s senior director of Regulatory Toxicology, Dr. Parker provides manufacturers with guidance on global regulatory and technical requirements and testing program design. In May 2019, Dr. Parker was appointed to a three-year term as co-chair of the Biological Evaluation (AAMI/BE) Committee, the U.S. mirror committee for ISO 10993. In addition, she is currently an internationally recognized ISO expert and a U.S. delegate for TC 194, the technical committee for ISO 10993, and is the past president of the Medical Device and Combination Products Specialty Section of the Society of Toxicology.
Dr. Ehman serves as a Technical and Regulatory director at WuXi AppTec Medical Device Testing, with a focus on medical device and combination products. Dr. Ehman has more than 18 years of toxicology and medical device experience, with expertise in toxicological risk assessments for medical devices, food and beverage products, and electronic nicotine delivery systems. In her current position, she provides medical device manufacturers with technical and regulatory support for biocompatibility test programs and conducts quantitative toxicological risk assessments to support product safety and risk management decisions.
The FDA's “Technical Considerations for Non-Clinical Assessment of Medical Devices Containing Nitinol” covers guidance in four primary areas: mechanical testing, corrosion, biocompatibility, and labeling. Identifying and resolving potential biocompatibility concerns when corrosion is a possibility can be challenging, especially with devices that were not previously required to meet this regulatory expectation. Standard chemical characterization testing cannot predict the potential toxicological effects of nickel ion release over time, which leaves a gap to fill during safety evaluation.
The Need for Nickel Ion Release Evaluations
The way the medical device industry evaluates products from a patient safety standpoint is constantly evolving. Nitinol differs from other metals that manufacturers might use in medical devices like stainless steel or titanium. Specifically, nitinol has shape memory qualities and flexibility that are difficult to replace with alternative materials.
The FDA’s guidance states, "Given the complex thermomechanical behavior of nitinol, there are unique considerations when assessing the safety and performance of nitinol devices."
Required methods that would traditionally identify material risks such as chemical characterization and toxicological risk assessments (TRAs) cannot fully capture its nickel release concerns. Therein, the need for alternative approaches remains. When a device has the potential for corrosion, and particularly when it is an implantable device, regulators are looking to see expanded testing to evaluate the risks of adverse health effects.
Nickel ion release is the primary concern with the presence of nitinol. Surface processing can have a significant impact on corrosion and nickel ion release. If a device does not have an established surface finish, manufacturers may need to perform an in-vitro nickel ion release study. Understanding the potential for nickel ion release may help the manufacturer optimize the manufacturing process if it decides to take a different approach with the surface finish of nitinol.
Key Call Outs in FDA Guidance Updates
Nitinol has long been a red flag for regulators evaluating cardiovascular devices, and with the increased interest in using minimally invasive procedures, nitinol applications have expanded.
Transcatheter heart valves, orthodontic devices, stents, and orthopedic fracture fixation (e.g., plates, screws, staples) are all examples of medical devices the FDA will be watching for evidence that nickel release is not a concern.
Manufacturers should evaluate any device containing nitinol that has either prolonged contact or long-term patient exposure under the latest guidance. Limited contact devices are considered at lower risk for nickel ion release and may not require the same testing. But any limited contact devices with an electrically active component, dissimilar metal couple or degradable metal/polymer components may also need to address nitinol risks under this guidance. In short, the test requirements for nickel ion release now include many more types of devices beyond just cardiovascular devices.
Manufacturers with nitinol-containing devices seeking advice on meeting regulatory expectations should consult the recent guidance. As shown by Figure 1, the addition of a corrosion testing flowchart acts as a valuable resource to understand expectations. This chart demonstrates where to look for corrosion testing acceptance criteria (ASTM F2129) and the steps to follow if testing does not meet criteria, or meets criteria but does not have an established surface finish. If additional testing is needed, manufacturers should use in-vitro nickel ion release methods to support the risk assessment.
Considerations for Prolonged and Long-Term Contacting Devices
Manufacturers can avoid costly delays by recognizing the pros and cons of each material choice before beginning safety testing. This foresight is crucial for prolonged or long-term contacting devices due to the heightened expectations. Even then, the slightest of changes to the manufacturing process can impact surface properties. These cases and any other corrosion possibility can affect a device's biocompatibility and the necessary testing requirements. If companies make manufacturing changes, consider additional testing and communicate goals with testing partners at the onset.
When laboratory testing partners have experience with nitinol and the testing needed, they can help avoid repeat studies and other potential delays. The investments in both biocompatibility and chemical characterization testing for implantable devices are substantial, and the last thing anyone wants is lack of communication to be the detrimental downfall.
The Limitations of Biocompatibility Testing Methods
Systemic toxicity is a concern for leachable metal ions, and while the FDA guidance document recommends an acceptable level for a TRA, this does not address sensitization concerns. The route and duration of exposure are key inputs to systemic toxicity; however, this endpoint can be addressed in a TRA because there is an established acceptable daily exposure.
Alternatively, sensitization is not easy to predict and functions in a manner much like an allergic reaction. The immune response leading to sensitization is variable between animal models and humans, and across the human population as a whole. Consequently, the use of different testing systems is not always predictive of the human response. Due to variability, there is no known lower limit on the amount of nickel that can elicit a sensitization response in some patients or be applied in the TRA.
The risk assessment of the chemical characterization and nickel ion release studies can address potential systemic toxicity to nickel. Still, regulators continue to view in-vivo models as the gold standard for addressing possible sensitization. Additionally, the guidance recommends potential risk mitigation through labeling, particularly for prolonged and long-term contacting devices.
Manufacturers who are still unsure how testing recommendations apply to their device can get feedback from regulators using a Q-sub process outlined in the guidance updates for direct consultation. The guidance also provides additional resources to help companies get started on this process.
Emerging Approaches to Protect Patient Safety
Protecting patient safety has taken new forms in recent years with a shift to more customized testing that addresses device-specific risks rather than a checklist approach. This guidance speaks to a clear theme of regulators looking into the potential material risks of each device. This trend is also evident in standards like ISO 10993-18:2020, which guides medical device biological safety testing through more detailed chemical characterization to support the evaluation of toxicological risks.
Additionally, a wide range of devices is now included under the standard, representing even more device categories and patient exposures than before. In particular, prolonged exposure and long-term contacting devices are the most affected by this guidance. For many companies, this might be a learning curve, but fortunately, they have resources including previous knowledge from the cardiovascular category and support from partners such as testing laboratories.
Before embarking on upcoming medical device safety testing, manufacturers should vet any potential impact from nitinol components on their test plan. Understanding the risks associated with each material choice is key, and customizing testing to fit this helps satisfy regulator questions.
Dr. Parker has more than 20 years of toxicology and medical device experience, and is an expert in biological evaluation of medical devices and combination products. She received her Ph.D. in molecular and cellular pharmacology from the University of Miami. In her current position as WuXi AppTec’s senior director of Regulatory Toxicology, Dr. Parker provides manufacturers with guidance on global regulatory and technical requirements and testing program design. In May 2019, Dr. Parker was appointed to a three-year term as co-chair of the Biological Evaluation (AAMI/BE) Committee, the U.S. mirror committee for ISO 10993. In addition, she is currently an internationally recognized ISO expert and a U.S. delegate for TC 194, the technical committee for ISO 10993, and is the past president of the Medical Device and Combination Products Specialty Section of the Society of Toxicology.
Dr. Ehman serves as a Technical and Regulatory director at WuXi AppTec Medical Device Testing, with a focus on medical device and combination products. Dr. Ehman has more than 18 years of toxicology and medical device experience, with expertise in toxicological risk assessments for medical devices, food and beverage products, and electronic nicotine delivery systems. In her current position, she provides medical device manufacturers with technical and regulatory support for biocompatibility test programs and conducts quantitative toxicological risk assessments to support product safety and risk management decisions.
