Dawn A. Lissy, President & Founder, Empirical05.19.17
For a company navigating a Class III medical device’s path to market, the U.S. Food and Drug Administration (FDA) panel presentation is the make-or-break point. That’s the day to present a case to convince the FDA the device—for which there’s no comparable predecessor—is safe and effective enough to be used on patients.
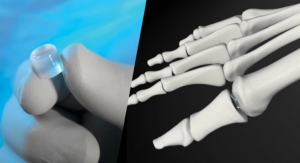
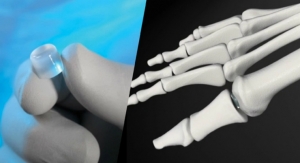
The buildup to that single day typically takes years. That panel presentation was nine years in the making for Cartiva Inc., a small, venture-backed medical device company. Its 20 employees banked Cartiva’s future on the approval of its initial product to market, the Cartiva Synthetic Cartilage Implant, the first implant of its kind to resurface the metatarsal joint.
“For your panel meeting, you have that one day,” said Deborah Moore, Cartiva’s vice president of clinical, regulatory, and quality affairs. “It’s eight hours. The future of your company is really made on that particular day. No matter how well we conducted that trial, no matter how good the data was, if we didn’t perform well on that day, it had a significant impact.”
Those nine years of diligent prep paid off when the implant earned approval in 71 days after the panel presentation. The average timespan for approval is 159 days.
Moore said finding the right partners to outsource aspects of the device’s development was a critical factor in that success.
“When we had a critical juncture, we outsourced those aspects,” she said. “The team here was really four individuals who were dedicated to the project. That’s not enough to get to the finish line.”
Moore contracted with Empirical Testing Corp. for mechanical testing. She turned to Hogan Lovells for legal and strategy support. For regulatory, clinical, and intense panel preparation, she worked with Musculoskeletal Clinical Regulatory Advisers LLC (MCRA). The Washington, D.C.-based full-service consulting firm specializes in musculoskeletal technology.
Glenn Stiegman, MCRA’s senior vice president of clinical and regulatory affairs, said the right partner knows the landscape of a client’s regulatory path, ideally down to the device’s specific body part and the surgeon installing the device.
“You want somebody that knows the anatomy and patient population, knows the study design that FDA would expect, knows that review group at the FDA—so you already have that established working relationship,” he said. “You have to understand the dynamics of the whole regulatory and clinical environment.”
Clients typically outsource to MCRA at three junctures, he said: beginning, middle, and in the wake of a catastrophic event. Cartiva reached out to MCRA in what Stiegman considers to be the middle.
The firm initially helped strategize responses to FDA’s questions about the device’s clinical study in a major deficiency letter. Over time, MCRA also provided clinical and regulatory strategy and support.
“We sort of jumped into the deep end since the PMA had been submitted,” Stiegman said. “We came in and said, ‘Let’s do it a different way.’ As time went on, we became more of the face of the company with the FDA.”
Eventually, Stiegman and other MCRA staffers also became the face of the FDA by playing the role of reviewers for two intense mock panel presentations it helped facilitate for Moore with Hogan Lovells.
“While a lot of time, strategy, and effort goes into preparing for panel, it’s still a crazy and intense time,” he said. “Being organized and understanding the questions the panel is asking, how best to respond, MCRA played a large role in that. We were working through each question, prepping [Moore] and others on how to respond, the sequence to do it in—MCRA was right by her side.”
Over the course of the two panel presentation test runs, Moore was grilled intensely. With the help of four more consultants preparing slides, pulling up data, and predicting the line of questioning, she presented her case to surgeons and consultants acting as FDA panel members.
“It’s one thing to sit in a conference and go back and forth, but to have the questions come from the clinicians at you in the same manner they’d be in the PMA meeting, that was absolutely critical,” she noted.
Moore said the actual panel presentation was less intense than her two practice runs. Although the mock panels added expenses to her budget, she said the money was “absolutely worth it.”
“We would do it again,” she said. “But I can see that amount of cost would [give] some companies pause.”
Finances are certainly a critical consideration for smaller companies, Stiegman said, but costs such as time to market, damage control, and generating sufficient data are also important factors.
“It’s a tough pill to swallow for a lot of smaller companies,” he said. “A lot of investors don’t get the fact that you have to look at the value creators and being successful at panel—ultimately success with the PMA is a major component, but not the only component. It’s sort of night and day when it comes to the success and the value creation, meaning, get through panel and PMA and you are done. When in reality, with PMA approval, you are just beginning.”
Not covering all possible bases can damn the device even if the FDA is satisfied with a panel presentation, Stiegman explained. Data required for reimbursement is a critical consideration that’s become an important factor in device development over the past 10-12 years, he said. It’s an aspect separate from premarket approval, but arguably just as critical to the product’s long-term success.
“The reimbursement part is sort of behind the curtain all along the process,” Stiegman said of reimbursement concerns on the path to market. “While ideally, the reimbursement data is there in the study from day one, most of the reimbursement data is never going to be reported in a panel setting. That doesn’t fit into safety/effectiveness concerns in [FDA] decision-making.”
When evaluating a consulting firm’s value, Stiegman advises clients to basically perform a self-audit. They must understand what will be necessary for regulatory approval, reimbursement success, and marketing success. Every device offers different challenges, but he said it comes down to a fundamental question: If cutting a particular expense adds risk to long-term success, is it worth it?
“If you don’t do it right, it’s more expensive in the long run,” Stiegman said. “You can spend triple the money going through appeals and going back to fix [earlier mistakes]. No one thanks you for saving money if you fail.”
Editor’s note: This is the final installment of a three-part series on bringing a Class III medical device to market. Part I, “David vs. Goliath: How a Small Startup Managed to Slingshot the Approval Process” and Part II, “Smart Outsourcing: Hiring the Team to Get Your Class III Device to Market” can be found in the November/December 2016 and January/February 2017 editions of ODT, respectively.
Dawn Lissy is a biomedical engineer, entrepreneur, and innovator. Since 1998, the Empirical family of companies (Empirical Testing Corp., Empirical Consulting LLC, and Empirical Machine LLC) has operated under Lissy’s direction. Empirical offers the full range of regulatory and quality systems consulting, testing, small batch and prototype manufacturing, and validations services to bring a medical device to market. Empirical is very active within standards development organization ASTM International and has one of the widest scopes of test methods of any accredited independent lab in the United States. Because Lissy was a member of the U.S. Food and Drug Administration’s Entrepreneur-in-Residence program, she has first-hand, in-depth knowledge of the regulatory landscape. Lissy holds an inventor patent for the Stackable Cage System for corpectomy and vertebrectomy. Her M.S. in biomedical engineering is from The University of Akron, Ohio.
The buildup to that single day typically takes years. That panel presentation was nine years in the making for Cartiva Inc., a small, venture-backed medical device company. Its 20 employees banked Cartiva’s future on the approval of its initial product to market, the Cartiva Synthetic Cartilage Implant, the first implant of its kind to resurface the metatarsal joint.
“For your panel meeting, you have that one day,” said Deborah Moore, Cartiva’s vice president of clinical, regulatory, and quality affairs. “It’s eight hours. The future of your company is really made on that particular day. No matter how well we conducted that trial, no matter how good the data was, if we didn’t perform well on that day, it had a significant impact.”
Those nine years of diligent prep paid off when the implant earned approval in 71 days after the panel presentation. The average timespan for approval is 159 days.
Moore said finding the right partners to outsource aspects of the device’s development was a critical factor in that success.
“When we had a critical juncture, we outsourced those aspects,” she said. “The team here was really four individuals who were dedicated to the project. That’s not enough to get to the finish line.”
Moore contracted with Empirical Testing Corp. for mechanical testing. She turned to Hogan Lovells for legal and strategy support. For regulatory, clinical, and intense panel preparation, she worked with Musculoskeletal Clinical Regulatory Advisers LLC (MCRA). The Washington, D.C.-based full-service consulting firm specializes in musculoskeletal technology.
Glenn Stiegman, MCRA’s senior vice president of clinical and regulatory affairs, said the right partner knows the landscape of a client’s regulatory path, ideally down to the device’s specific body part and the surgeon installing the device.
“You want somebody that knows the anatomy and patient population, knows the study design that FDA would expect, knows that review group at the FDA—so you already have that established working relationship,” he said. “You have to understand the dynamics of the whole regulatory and clinical environment.”
Clients typically outsource to MCRA at three junctures, he said: beginning, middle, and in the wake of a catastrophic event. Cartiva reached out to MCRA in what Stiegman considers to be the middle.
The firm initially helped strategize responses to FDA’s questions about the device’s clinical study in a major deficiency letter. Over time, MCRA also provided clinical and regulatory strategy and support.
“We sort of jumped into the deep end since the PMA had been submitted,” Stiegman said. “We came in and said, ‘Let’s do it a different way.’ As time went on, we became more of the face of the company with the FDA.”
Eventually, Stiegman and other MCRA staffers also became the face of the FDA by playing the role of reviewers for two intense mock panel presentations it helped facilitate for Moore with Hogan Lovells.
“While a lot of time, strategy, and effort goes into preparing for panel, it’s still a crazy and intense time,” he said. “Being organized and understanding the questions the panel is asking, how best to respond, MCRA played a large role in that. We were working through each question, prepping [Moore] and others on how to respond, the sequence to do it in—MCRA was right by her side.”
Over the course of the two panel presentation test runs, Moore was grilled intensely. With the help of four more consultants preparing slides, pulling up data, and predicting the line of questioning, she presented her case to surgeons and consultants acting as FDA panel members.
“It’s one thing to sit in a conference and go back and forth, but to have the questions come from the clinicians at you in the same manner they’d be in the PMA meeting, that was absolutely critical,” she noted.
Moore said the actual panel presentation was less intense than her two practice runs. Although the mock panels added expenses to her budget, she said the money was “absolutely worth it.”
“We would do it again,” she said. “But I can see that amount of cost would [give] some companies pause.”
Finances are certainly a critical consideration for smaller companies, Stiegman said, but costs such as time to market, damage control, and generating sufficient data are also important factors.
“It’s a tough pill to swallow for a lot of smaller companies,” he said. “A lot of investors don’t get the fact that you have to look at the value creators and being successful at panel—ultimately success with the PMA is a major component, but not the only component. It’s sort of night and day when it comes to the success and the value creation, meaning, get through panel and PMA and you are done. When in reality, with PMA approval, you are just beginning.”
Not covering all possible bases can damn the device even if the FDA is satisfied with a panel presentation, Stiegman explained. Data required for reimbursement is a critical consideration that’s become an important factor in device development over the past 10-12 years, he said. It’s an aspect separate from premarket approval, but arguably just as critical to the product’s long-term success.
“The reimbursement part is sort of behind the curtain all along the process,” Stiegman said of reimbursement concerns on the path to market. “While ideally, the reimbursement data is there in the study from day one, most of the reimbursement data is never going to be reported in a panel setting. That doesn’t fit into safety/effectiveness concerns in [FDA] decision-making.”
When evaluating a consulting firm’s value, Stiegman advises clients to basically perform a self-audit. They must understand what will be necessary for regulatory approval, reimbursement success, and marketing success. Every device offers different challenges, but he said it comes down to a fundamental question: If cutting a particular expense adds risk to long-term success, is it worth it?
“If you don’t do it right, it’s more expensive in the long run,” Stiegman said. “You can spend triple the money going through appeals and going back to fix [earlier mistakes]. No one thanks you for saving money if you fail.”
Editor’s note: This is the final installment of a three-part series on bringing a Class III medical device to market. Part I, “David vs. Goliath: How a Small Startup Managed to Slingshot the Approval Process” and Part II, “Smart Outsourcing: Hiring the Team to Get Your Class III Device to Market” can be found in the November/December 2016 and January/February 2017 editions of ODT, respectively.
Dawn Lissy is a biomedical engineer, entrepreneur, and innovator. Since 1998, the Empirical family of companies (Empirical Testing Corp., Empirical Consulting LLC, and Empirical Machine LLC) has operated under Lissy’s direction. Empirical offers the full range of regulatory and quality systems consulting, testing, small batch and prototype manufacturing, and validations services to bring a medical device to market. Empirical is very active within standards development organization ASTM International and has one of the widest scopes of test methods of any accredited independent lab in the United States. Because Lissy was a member of the U.S. Food and Drug Administration’s Entrepreneur-in-Residence program, she has first-hand, in-depth knowledge of the regulatory landscape. Lissy holds an inventor patent for the Stackable Cage System for corpectomy and vertebrectomy. Her M.S. in biomedical engineering is from The University of Akron, Ohio.