Sam Brusco, Associate Editor02.23.17
Tropicana never expected a simple packaging change would so severely sour its customers.
In 2009, the company altered the design of its orange juice cartons to a more modern image by swapping out its traditional straw-in-orange logo with a glass of orange juice. Unfortunately, after doing so, they lost about a fifth of their sales in a few weeks, according to Business Insider. Consumers were so infuriated by the switch they complained about the redesign in letters, emails, and telephone calls, begging for a return to the original logo. According to an article on the subject in The New York Times, the new design was described as “ugly,” “stupid,” and resembling “a generic bargain brand.” Tropicana loyalists apparently set store by the freshness evoked in that straw in the orange, and based on the intensity of the outcry, felt personally attacked by the change. “Tropicana has something like eight feet in the refrigerated section. People recognized it. They didn't trust [the new logo],” Olson chief creative officer Dennis Ryan told Business Insider. Thankfully, the company quickly resolved the issue and brought back the original design, restoring order in the citrus kingdom.
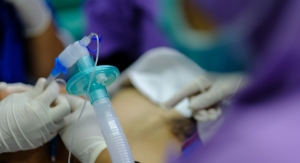
A simple logo change wouldn’t necessarily elicit such strong response from orthopedic device customers, but packaging design still has a significant impact on a musculoskeletal device or implant’s efficacy. The primary function of packaging (apart from branding) is to keep the device free from contamination during transport. Orthopedic implants in particular must remain sterile from manufacture through implantation to avoid messy post-op complications. When designing an orthopedic device, it would be prudent to keep its packaging and sterilization methods in mind early in development to avert any contamination crises resulting in recalls. However, some orthopedic OEMs aren’t knowledgeable enough about the process, or otherwise lack the resources to pay such strong detail to keeping a device clean.
In order to gain insight on the trends and challenges impacting orthopedic device packaging and sterilization, seven participants involved in various areas across the orthopedic device packaging and sterilization sector spoke with ODT:
Seán Egan: Packaging must protect the device, maintaining sterile barrier integrity to the point of use. It must also enable medical staff to clearly identify the part and confirm it has maintained its barrier properties, with no punctures or abrasion damage to the pack or part in transport. Additionally, packaging needs to eliminate costs through reduction of waste in material, storage, and transportation.
Wendy Mach: In 2016, we saw a few companies struggle with incorrect labeling and problematic packaging. One example that stands out in was that a particular orthopedic device manufacturer had a recall on an LDPE bag where essentially, the outer surface wore away, creating possible particulate matter within the package. Several manufacturers had identified packages that were possibly not sealed.
Gary Socola: With little warning to the manufacturers themselves, it’s difficult for orthopedic device or implant OEMs to stay current with all of the U.S. Food and Drug Administration (FDA) policy and procedural changes. OEMs typically find out about these changes only after submitting a device for clearance, and the FDA notifies them about new changes and the agency’s new line of thinking. It can be difficult to form a strategy of device validation when the rules of the game are constantly changing. We recommend that many of our clients should contact the FDA before submission to clarify any areas of concern—or to even file a pre-510(k) submission if the device is different enough or has significantly different labeling claims than prior cleared devices under the same FDA product code.
Brian Whalen: Medical devices and their corresponding accessories are becoming increasingly complex and sophisticated while being transported quite literally across the globe, making it more of a challenge to ensure the device arrives safely and securely to the patient.
Ben White: FDA and ISO standards continue to become increasingly stringent and quality critical. For OEMs, the most pressing issue resides in how to abide by the standards, as they can occasionally be left to interpretation. With the evolving regulation on endotoxin batch testing, we are seeing OEMs tackle requirements differently and some are unsure what the best solution is.
Brusco: Do you find orthopedic device OEMs view packaging as an important part of design? If not, what benefits can be reaped by considering package design early in the process?
Egan: As devices become more sophisticated, it is becoming increasingly important to look at packaging design earlier in the development process. Packaging can enhance the user experience by meeting the surgical team’s needs in the preparation and delivery of the device to the patient. Use of human factors in both device and packaging design helps reduce risk by guiding the clinician through the necessary steps to deliver treatment. For many orthopedic devices, new materials/coatings on the device present unique challenges for packaging to protect the device in direct contact with the packaging material, particularly against scuffing of highly-finished surfaces—which can impact product life.
Mach: I think that this is a focus in the industry and becoming more important, but there are still clients that look at packaging as just a byproduct of their device. They are not prepared to deal with the consequences (capital or time) when they fail their validations.
Mason Schwartz: Yes, in some aspects—some put more depth of knowledge and thought into this aspect, but others just see it as a necessary evil, rather than an essential part of the device, instead utilizing more depth of knowledge and thought in some areas. The 50 percent who do care about the packaging can come up with a more aesthetically pleasing and functional package for the end user. An engineer also needs to keep the sterilization modality in mind when designing their package.
Whalen: Oftentimes, OEMs don’t consider packaging early enough in the product development process. By engaging in packaging design early, many headaches can be avoided down the road. When packaging is rushed through development at the eleventh hour, the chance of mistakes and failures greatly increase, which can drive costs up and delay product launch. When this critical process is considered early in development, proper testing protocols are created and executed, user feedback is considered and implemented, and a cost-effective packing solution is created.
White: Often, the number one goal for OEMs is getting a product to market as fast as possible. The easiest way to accomplish that is to sacrifice the packaging design portion of product development, as long as package integrity is maintained. There is a delicate balance between taking the time to create a robust package design and increasing your time to market. Up to 12-month delays can occur due to insufficient allocation of time upfront. Failure to dedicate the appropriate time and resources during this stage of product design can create significant setbacks during validations. A robust package design prevents failures during the validation stage and while the product is in the field.
Brusco: How can packaging help alert when the sterile barrier for an orthopedic device has been breached?
Egan: Training of clinical staff to identify breaches in the seal area of tray lid combinations is still pretty much the first line of defense to check that integrity has not been compromised. Use of color on the seal flange can aid visual inspection of the seal area to highlight issues which might indicate a breach. A blue tint is common in many trays which appear darker when sealed against lidding material; light patches in this seal can be an indication of a void or breach of the barrier layer.
Mach: This is self-explanatory. If the package is damaged in any manner, it should be considered breached until they have information from the manufacturer that says otherwise.
Socola: I don’t believe there is anything better than an inspection by the healthcare user to pick up any breaches in a packaging system. Procedures for these inspections are described in the American National Standard AAMI ST-79, the AORN guidelines, and by the packaging system manufacturer. However, companies like Halyard Health are trying. According to Halyard, their SMART-FOLD sterilization wrap is ‘less likely, but, if a breach has occurred, the contrasting white inner fabric layer makes it easy to check for any tears or cuts.’ Cardinal Health has their Dual Layer Sterilization Wrap with one blue and one green sheet. According to Cardinal Health, their customers only need to select the color they want (blue or green) to help differentiate, identify, and inspect trays.
White: Design. It is important to consider package design up-front and develop confidence during the validation stage. This stage of product development assures the packaged device can withstand all subjected variables and reach its final destination while maintaining the integrity of the sterile barrier.
Brusco: How have sterilization processes for orthopedic devices evolved in the last few years? Have they remained stagnant? If so, why?
Janet Prust: For reusable medical/orthopedic devices, the key evolution is reduction of non-FDA cleared “extended cycles” processes. Extended cycles are non-traditional saturated steam cycles used in hospitals to sterilize reusable devices, which were too frequently recommended for large, heavy sets. This was due to the large metal mass and complex lumen requirements to achieve sterilization. These cycles created a significant burden and challenge for healthcare facilities, and while based on acceptable sterilization validations by the device manufacture, the extended cycles were not available in FDA-cleared healthcare steam sterilizers. This created off-label use by the facility, which included risk of selecting the wrong cycle. Device manufacturers have responded to this by reducing set complexity, and in many cases, completing new sterilization validations and FDA clearances. Extended cycles are no longer cleared by FDA in the device instructions for use, but many legacy products still require the facility to use these cycles. More redesign and revalidation work is needed to eliminate the need for extended cycles.
Schwartz: Regulations generally present a high barrier to development and implementation of new sterilization methods, but more new viable methods have emerged over the last two to five years than in the previous 20. In the U.S., the controlling FDA guidance document (released in early 2016) classifies sterilization methods according to three categories. REVOX VPA is currently categorized as a “Novel Sterilization” method, but the burden of proof of efficacy and safety is the same as with any other method. While the sterilization techniques in this category do not yet possess a history of comprehensive FDA evaluations, REVOX has been approved for use as the sterilization method on a FDA 510(k) Class II implantable device. This represents the first FDA-cleared device with the use of a novel sterilization method in more than 20 years.
Socola: By all means, they have not remained stagnant! Hospitals have more options for complex device sterilization than ever before. The FDA continues to clear new low temperature sterilization processes for use in the U.S. healthcare market and OEMs are doing their best to keep up with these new processes by having their devices validated in them. To cite a few specific examples, Advanced Sterilization Products (ASP), manufacturer of the STERRAD line of hydrogen peroxide (H2O2) sterilizers, received a new 510(k) clearance that allows a hospital’s IT department to connect some of their devices to the hospital’s local area network for transfer of cycle parameters to a server and then, if desired, to an instrument tracking system. They followed this up with additional FDA clearances in September, which paved the way for their new ALLClear Technology. This technology, according to ASP, incorporates enhancements and new features to improve reliability and usability. TSO3 Inc. also received a new 510(k) clearance in June 2016 for their H2O2/ozone sterilization process. According to TSO3, it offers a single sterilization cycle intended for general instruments, flexible endoscopes (including single, dual, and multi-channel devices), and rigid-channel devices (including single-channel and double channel rigid endoscopes) and the unit can process loads of up to 75 lbs. STERIS also added new claims in July of 2016 to their line of H2O2 low-temperature sterilizers. According to STERIS, the new FDA clearance is to modify the maximum load weight for the Non Lumen Cycle from 19.65 lbs to 50 lbs, to add titanium diffusion-restricted areas to the indications for use of the Non Lumen Cycle, and to remove the term “reusable” from their indications for use statement.
Brusco: What challenges arise in developing sterilization methods for orthopedic devices containing innovative materials, embedded electronics, or drug-device combination products?
Prust: Challenges relating to materials compatibility, sterilant penetration into complex geometries, and cycle time directly impacts manufacturing productivity and ultimately, profitability. New materials and designs can require customized cycles specific to the single-use device. Our historical approach of designing new devices that fit into existing families and cycles may no longer be possible with new innovations.
Embedded electronics are a challenge for certain types of processes. Current compliant mechanical design in the sterilization systems is important to ensure intrinsically safe hardware for some sterilization processes used to sterilize electronic components. Old systems built under outdated safety regulations may not be appropriate for new electronic designs. All sterilization processes must be not only efficacious, but safe for both the operator of the sterilization system and the patient receiving that device. Not all processes are compatible with embedded electronics, and as innovation in this area continues, optimization of these safe processes will need to continue. As for combination products, the pharmacological or therapeutic agent activity can be affected by the sterilization process or cycle parameters. Optimized cycles specific to the combination device will be required more frequently as they become more complex. As new combination products are designed and sterilization validations are conducted, some sterilization technologies, such as ethylene oxide (EtO), are available for easy customization by the device manufacturer.
Schwartz: Combination devices pose an interesting challenge for sterilization and newer novel sterilization methods are being reviewed, as traditional sterilization methods often fall short of meeting the Sterilator standards as well as product quality and efficacy requirements. Electronic devices have issues with radiation (depending on circuitry), and standard sterilization technologies—such as heat and radiation—will interfere with materials, whereas novel methods may offer better results.
Socola: Two issues that come to mind are the biocompatibility of the material with the sterilization process itself (i.e., the material can’t be toxic or emit any harmful byproducts) and the functionality of the material itself, after being exposed to multiple sterilization cycles and in multiple cleaning processes. We have performed reusability studies where materials were subjected to hundreds of sterilization cycles in steam, EtO, and H2O2 sterilization processes and depending on the material, many times degradation will appear in one sterilization process much sooner than it does in another. For instance, materials sensitive to EtO may break down after 20 cycles, but the same material in steam may look untouched after 500 steam cycles.
A methodology to determine the worst-case component or material in the combination product may need to be developed by the OEM, in conjunction with their chosen test laboratory. This information, along with the determination of the worst-case locations for sterilant penetration, would be used to develop an appropriate sterilization efficacy validation which would result in the appropriate sterility assurance level (SAL) of 10-6.
White: Consideration to the product and its material composition, as well as the packaging configuration’s material and design, is essential. The end goal is to remove microorganisms to avoid affecting the product in a negative manner; this poses a unique challenge when working with innovative biomaterials.
Brusco: Can you offer insight on Unique Device Identification (UDI) and its impact on packaging requirements?
Egan: The effects of UDI on packaging still has not filtered down to thermoformers in general, as most of the requirements come into effect after the trays and blisters have been delivered to the OEM.
Socola: We can only offer insight from the perspective of a validation lab. The reprocessing of an orthopedic device multiple times (i.e., clean and disinfect/sterilize) can affect the legibility and function of any labeling on the device itself. Therefore, OEMs should consider materials compatibility testing as part of design validation for any device that has the UDI directly on it to ensure that after multiple uses, this information is still legible and no adverse defects are noted. We have seen degradation to the UDI on devices in our lab after multiple exposures to the cleaning and sterilization process.
Whalen: The FDA has released a final ruling requiring that most medical devices distributed in the United States carry a unique device identifier, or UDI. It also applies to certain combination products that contain devices and those licensed under the Public Health Service. Compliance dates depend on classification of the device. Class III devices have the earliest requirement dates. A UDI system has the potential to improve the quality of information in medical device adverse event reports, which will help the FDA identify product problems more quickly, better target recalls, and improve patient safety. CleanCut is well positioned to support our customers in helping them meet these requirements with our various labeling and printing systems and equipment.
White: For OEMs, the amount of up-front work surrounding registration of devices, GTIN barcoding, overall label validation, and the potential impact on field inventory for rework and re-label as compliance deadlines approach can be cumbersome. Partnering with a packaging supplier to assist with the transition to serialized inventory will be an effective option for OEMs.
Brusco: What changes to orthopedic device packaging and sterilization do you see happening in the years to come?
Egan: Medical device manufacturers will continue to drive efficiencies in their processes to reduce costs. Packaging design must protect the device to point-of-use but needs to take into consideration “total cost of ownership” to the OEM through efficient design that eliminates material waste, reduces transport/storage costs and sterilization costs. Packaging will need to add functionality to the device and procedure by meeting the needs of the surgical team. For example, presenting all components required for the procedure in one handling tray or in aiding the clinician in prepping the device before introducing it to the patient.
Mach: There is a big push for recycling medical packaging right now. Although it’s in its early stages, in the future we will see large strides by manufacturers to designate materials as recyclable, and hospitals working toward recycling those materials—bringing about less impact on landfills.
Prust: Single-use device packaging will continue to become “greener” by reducing non-sustainable materials and dimensions. Combination devices are often humidity and temperature sensitive, which in some applications can be addressed by specific packaging design and component materials to help protect the product during transportation. Reusable device packaging will continue to migrate to containerized systems specific to the device with improved sealing and integrity detection systems.
Schwartz: In the future, we will continue to see greater need for faster, more cost-effective, and more nimble sterilization methods. For example, patient-specific implants manufactured with 3D printing need to be built and sterilized quickly and close to where the procedure will occur. Current sterilization methods rely on large batch manufacturing processes that don't fit that fast-turn model. Sterilization methods of the future will be scaled to both ends of the throughput volume requirement spectrum, enable in-line processing for quick turns and greater manufacturing efficiency, can be performed with parametric release, and offer a fast feasibility-to-validation process. We will also see the possibility for alternative packaging materials to Tyvek, as well as the ability to incorporate novel component materials, such as with combination devices.
Socola: For reusable orthopedic devices/sets processed within healthcare facilities, the packaging has evolved in recent times from wrapping the tray/set in two layers of a sterilization wrapper before sterilization, to placing the tray or set within a rigid sterilization container. Orthopedic manufacturers may recommend this practice for a number of reasons. One reason stated by the OEM is the weight of the tray or set itself, as many orthopedic trays/sets are fairly heavy and may cause some sterilization wraps to be breached or compromised by either abrasion (placed on and off shelves) or through compression (as when stacking sets on top of one another in sterile storage). Under these situations, the use of rigid sterilization containers may aid in the stacking and removal of trays and sets from storage shelves while helping to maintain package integrity. However, there have been significant changes to sterilization wraps in recent years as well, and this trend is likely to continue. There are now more companies selling SMS sterilization wrap in the North American healthcare market than ever before.
Whalen: More hospitals are now requesting that orthopedic medical device makers have their implants arrive individually sterilized. In the past, hospitals would autoclave entire trays of non-sterile implants prior to surgery. This practice, however, raises questions about the integrity of the implant following repeated sterilization cycles, a lack of traceability of implant batch, and most significantly, the correlation to post-operative infection and its costly consequences.
White: There will be an increase in the utilization of a single package configuration across multiple product designs. This provides a type of “pre-validated packaging” to increase time to market while reducing upfront costs. Single package designs that fit a vast majority of devices will help reduce the environmental footprint of packaging. Additionally, improved packaging equipment and methods will be vital for OEMs to improve costs and associated lead times.
Brusco: Is there anything else regarding orthopedic device packaging and/or sterilization you'd like to say?
Schwartz: Universal sterilization requirements and standards across the world would drive simpler regulatory approvals. This would be a vast improvement over each country having their own set of requirements.
Socola: OEMs can help themselves tremendously by putting together validation teams during product development and before device testing. I can’t tell you the number of times we received an orthopedic set for validation testing to only find out that the silicon tabs which tightly hold a device in place within the sterilization set are actually impeding sterilant penetration. The number of access holes in the tray itself and vent to volumes ratios also can have a dramatic effect of device sterilization. If these design issues are not addressed early enough, they can lead to sterilization efficacy failures and ultimately a redesign of the tray or set itself in one way or another. It most cases, design engineers working with experts in sterilization can make design changes early on in the process to significantly aid in getting the desired results during sterilization efficacy validation studies.
Whalen: Almost as important as the OEM medical or orthopedic device is its packaging. Today’s packaging is designed to be as unique and effective as the device itself. Much needs to be considered in the process. The packaging solution must maintain the sterile barrier system, protect the device in transit and use, and successfully communicate the OEM’s branding message. When this process is considered early, costly mistakes can be avoided and the ideal packaging solution can be created.
White: Allocating sufficient time for packaging and sterilization validations is essential to get products to market effectively and efficiently. Proper execution provides assurance of product quality and patient safety.
In 2009, the company altered the design of its orange juice cartons to a more modern image by swapping out its traditional straw-in-orange logo with a glass of orange juice. Unfortunately, after doing so, they lost about a fifth of their sales in a few weeks, according to Business Insider. Consumers were so infuriated by the switch they complained about the redesign in letters, emails, and telephone calls, begging for a return to the original logo. According to an article on the subject in The New York Times, the new design was described as “ugly,” “stupid,” and resembling “a generic bargain brand.” Tropicana loyalists apparently set store by the freshness evoked in that straw in the orange, and based on the intensity of the outcry, felt personally attacked by the change. “Tropicana has something like eight feet in the refrigerated section. People recognized it. They didn't trust [the new logo],” Olson chief creative officer Dennis Ryan told Business Insider. Thankfully, the company quickly resolved the issue and brought back the original design, restoring order in the citrus kingdom.
A simple logo change wouldn’t necessarily elicit such strong response from orthopedic device customers, but packaging design still has a significant impact on a musculoskeletal device or implant’s efficacy. The primary function of packaging (apart from branding) is to keep the device free from contamination during transport. Orthopedic implants in particular must remain sterile from manufacture through implantation to avoid messy post-op complications. When designing an orthopedic device, it would be prudent to keep its packaging and sterilization methods in mind early in development to avert any contamination crises resulting in recalls. However, some orthopedic OEMs aren’t knowledgeable enough about the process, or otherwise lack the resources to pay such strong detail to keeping a device clean.
In order to gain insight on the trends and challenges impacting orthopedic device packaging and sterilization, seven participants involved in various areas across the orthopedic device packaging and sterilization sector spoke with ODT:
- Seán Egan, group marketing manager of Cranston, R.I.- based Nelipak Healthcare Packaging
- Wendy Mach, packaging section leader of Salt Lake City, Utah-based Nelson Laboratories
- Janet Prust, director of standards and global business development of St. Paul, Minn.-based 3M’s Infection Division
- Mason Schwartz, director of R&D and operations of Minneapolis, Minn.-based REVOX Sterilization Solutions, a division of Cantel Medical Corp.
- Gary Socola, president of Rochester, N.Y.-based HIGHPOWER Validation Testing & Lab Services Inc.
- Brian Whalen, director of sales and marketing of Anaheim, Calif.-based CleanCut Technologies
- Ben White, engineering manager, business development of Fall River, Mass.-based Millstone Medical Outsourcing
Seán Egan: Packaging must protect the device, maintaining sterile barrier integrity to the point of use. It must also enable medical staff to clearly identify the part and confirm it has maintained its barrier properties, with no punctures or abrasion damage to the pack or part in transport. Additionally, packaging needs to eliminate costs through reduction of waste in material, storage, and transportation.
Wendy Mach: In 2016, we saw a few companies struggle with incorrect labeling and problematic packaging. One example that stands out in was that a particular orthopedic device manufacturer had a recall on an LDPE bag where essentially, the outer surface wore away, creating possible particulate matter within the package. Several manufacturers had identified packages that were possibly not sealed.
Gary Socola: With little warning to the manufacturers themselves, it’s difficult for orthopedic device or implant OEMs to stay current with all of the U.S. Food and Drug Administration (FDA) policy and procedural changes. OEMs typically find out about these changes only after submitting a device for clearance, and the FDA notifies them about new changes and the agency’s new line of thinking. It can be difficult to form a strategy of device validation when the rules of the game are constantly changing. We recommend that many of our clients should contact the FDA before submission to clarify any areas of concern—or to even file a pre-510(k) submission if the device is different enough or has significantly different labeling claims than prior cleared devices under the same FDA product code.
Brian Whalen: Medical devices and their corresponding accessories are becoming increasingly complex and sophisticated while being transported quite literally across the globe, making it more of a challenge to ensure the device arrives safely and securely to the patient.
Ben White: FDA and ISO standards continue to become increasingly stringent and quality critical. For OEMs, the most pressing issue resides in how to abide by the standards, as they can occasionally be left to interpretation. With the evolving regulation on endotoxin batch testing, we are seeing OEMs tackle requirements differently and some are unsure what the best solution is.
Brusco: Do you find orthopedic device OEMs view packaging as an important part of design? If not, what benefits can be reaped by considering package design early in the process?
Egan: As devices become more sophisticated, it is becoming increasingly important to look at packaging design earlier in the development process. Packaging can enhance the user experience by meeting the surgical team’s needs in the preparation and delivery of the device to the patient. Use of human factors in both device and packaging design helps reduce risk by guiding the clinician through the necessary steps to deliver treatment. For many orthopedic devices, new materials/coatings on the device present unique challenges for packaging to protect the device in direct contact with the packaging material, particularly against scuffing of highly-finished surfaces—which can impact product life.
Mach: I think that this is a focus in the industry and becoming more important, but there are still clients that look at packaging as just a byproduct of their device. They are not prepared to deal with the consequences (capital or time) when they fail their validations.
Mason Schwartz: Yes, in some aspects—some put more depth of knowledge and thought into this aspect, but others just see it as a necessary evil, rather than an essential part of the device, instead utilizing more depth of knowledge and thought in some areas. The 50 percent who do care about the packaging can come up with a more aesthetically pleasing and functional package for the end user. An engineer also needs to keep the sterilization modality in mind when designing their package.
Whalen: Oftentimes, OEMs don’t consider packaging early enough in the product development process. By engaging in packaging design early, many headaches can be avoided down the road. When packaging is rushed through development at the eleventh hour, the chance of mistakes and failures greatly increase, which can drive costs up and delay product launch. When this critical process is considered early in development, proper testing protocols are created and executed, user feedback is considered and implemented, and a cost-effective packing solution is created.
White: Often, the number one goal for OEMs is getting a product to market as fast as possible. The easiest way to accomplish that is to sacrifice the packaging design portion of product development, as long as package integrity is maintained. There is a delicate balance between taking the time to create a robust package design and increasing your time to market. Up to 12-month delays can occur due to insufficient allocation of time upfront. Failure to dedicate the appropriate time and resources during this stage of product design can create significant setbacks during validations. A robust package design prevents failures during the validation stage and while the product is in the field.
Brusco: How can packaging help alert when the sterile barrier for an orthopedic device has been breached?
Egan: Training of clinical staff to identify breaches in the seal area of tray lid combinations is still pretty much the first line of defense to check that integrity has not been compromised. Use of color on the seal flange can aid visual inspection of the seal area to highlight issues which might indicate a breach. A blue tint is common in many trays which appear darker when sealed against lidding material; light patches in this seal can be an indication of a void or breach of the barrier layer.
Mach: This is self-explanatory. If the package is damaged in any manner, it should be considered breached until they have information from the manufacturer that says otherwise.
Socola: I don’t believe there is anything better than an inspection by the healthcare user to pick up any breaches in a packaging system. Procedures for these inspections are described in the American National Standard AAMI ST-79, the AORN guidelines, and by the packaging system manufacturer. However, companies like Halyard Health are trying. According to Halyard, their SMART-FOLD sterilization wrap is ‘less likely, but, if a breach has occurred, the contrasting white inner fabric layer makes it easy to check for any tears or cuts.’ Cardinal Health has their Dual Layer Sterilization Wrap with one blue and one green sheet. According to Cardinal Health, their customers only need to select the color they want (blue or green) to help differentiate, identify, and inspect trays.
White: Design. It is important to consider package design up-front and develop confidence during the validation stage. This stage of product development assures the packaged device can withstand all subjected variables and reach its final destination while maintaining the integrity of the sterile barrier.
Brusco: How have sterilization processes for orthopedic devices evolved in the last few years? Have they remained stagnant? If so, why?
Janet Prust: For reusable medical/orthopedic devices, the key evolution is reduction of non-FDA cleared “extended cycles” processes. Extended cycles are non-traditional saturated steam cycles used in hospitals to sterilize reusable devices, which were too frequently recommended for large, heavy sets. This was due to the large metal mass and complex lumen requirements to achieve sterilization. These cycles created a significant burden and challenge for healthcare facilities, and while based on acceptable sterilization validations by the device manufacture, the extended cycles were not available in FDA-cleared healthcare steam sterilizers. This created off-label use by the facility, which included risk of selecting the wrong cycle. Device manufacturers have responded to this by reducing set complexity, and in many cases, completing new sterilization validations and FDA clearances. Extended cycles are no longer cleared by FDA in the device instructions for use, but many legacy products still require the facility to use these cycles. More redesign and revalidation work is needed to eliminate the need for extended cycles.
Schwartz: Regulations generally present a high barrier to development and implementation of new sterilization methods, but more new viable methods have emerged over the last two to five years than in the previous 20. In the U.S., the controlling FDA guidance document (released in early 2016) classifies sterilization methods according to three categories. REVOX VPA is currently categorized as a “Novel Sterilization” method, but the burden of proof of efficacy and safety is the same as with any other method. While the sterilization techniques in this category do not yet possess a history of comprehensive FDA evaluations, REVOX has been approved for use as the sterilization method on a FDA 510(k) Class II implantable device. This represents the first FDA-cleared device with the use of a novel sterilization method in more than 20 years.
Socola: By all means, they have not remained stagnant! Hospitals have more options for complex device sterilization than ever before. The FDA continues to clear new low temperature sterilization processes for use in the U.S. healthcare market and OEMs are doing their best to keep up with these new processes by having their devices validated in them. To cite a few specific examples, Advanced Sterilization Products (ASP), manufacturer of the STERRAD line of hydrogen peroxide (H2O2) sterilizers, received a new 510(k) clearance that allows a hospital’s IT department to connect some of their devices to the hospital’s local area network for transfer of cycle parameters to a server and then, if desired, to an instrument tracking system. They followed this up with additional FDA clearances in September, which paved the way for their new ALLClear Technology. This technology, according to ASP, incorporates enhancements and new features to improve reliability and usability. TSO3 Inc. also received a new 510(k) clearance in June 2016 for their H2O2/ozone sterilization process. According to TSO3, it offers a single sterilization cycle intended for general instruments, flexible endoscopes (including single, dual, and multi-channel devices), and rigid-channel devices (including single-channel and double channel rigid endoscopes) and the unit can process loads of up to 75 lbs. STERIS also added new claims in July of 2016 to their line of H2O2 low-temperature sterilizers. According to STERIS, the new FDA clearance is to modify the maximum load weight for the Non Lumen Cycle from 19.65 lbs to 50 lbs, to add titanium diffusion-restricted areas to the indications for use of the Non Lumen Cycle, and to remove the term “reusable” from their indications for use statement.
Brusco: What challenges arise in developing sterilization methods for orthopedic devices containing innovative materials, embedded electronics, or drug-device combination products?
Prust: Challenges relating to materials compatibility, sterilant penetration into complex geometries, and cycle time directly impacts manufacturing productivity and ultimately, profitability. New materials and designs can require customized cycles specific to the single-use device. Our historical approach of designing new devices that fit into existing families and cycles may no longer be possible with new innovations.
Embedded electronics are a challenge for certain types of processes. Current compliant mechanical design in the sterilization systems is important to ensure intrinsically safe hardware for some sterilization processes used to sterilize electronic components. Old systems built under outdated safety regulations may not be appropriate for new electronic designs. All sterilization processes must be not only efficacious, but safe for both the operator of the sterilization system and the patient receiving that device. Not all processes are compatible with embedded electronics, and as innovation in this area continues, optimization of these safe processes will need to continue. As for combination products, the pharmacological or therapeutic agent activity can be affected by the sterilization process or cycle parameters. Optimized cycles specific to the combination device will be required more frequently as they become more complex. As new combination products are designed and sterilization validations are conducted, some sterilization technologies, such as ethylene oxide (EtO), are available for easy customization by the device manufacturer.
Schwartz: Combination devices pose an interesting challenge for sterilization and newer novel sterilization methods are being reviewed, as traditional sterilization methods often fall short of meeting the Sterilator standards as well as product quality and efficacy requirements. Electronic devices have issues with radiation (depending on circuitry), and standard sterilization technologies—such as heat and radiation—will interfere with materials, whereas novel methods may offer better results.
Socola: Two issues that come to mind are the biocompatibility of the material with the sterilization process itself (i.e., the material can’t be toxic or emit any harmful byproducts) and the functionality of the material itself, after being exposed to multiple sterilization cycles and in multiple cleaning processes. We have performed reusability studies where materials were subjected to hundreds of sterilization cycles in steam, EtO, and H2O2 sterilization processes and depending on the material, many times degradation will appear in one sterilization process much sooner than it does in another. For instance, materials sensitive to EtO may break down after 20 cycles, but the same material in steam may look untouched after 500 steam cycles.
A methodology to determine the worst-case component or material in the combination product may need to be developed by the OEM, in conjunction with their chosen test laboratory. This information, along with the determination of the worst-case locations for sterilant penetration, would be used to develop an appropriate sterilization efficacy validation which would result in the appropriate sterility assurance level (SAL) of 10-6.
White: Consideration to the product and its material composition, as well as the packaging configuration’s material and design, is essential. The end goal is to remove microorganisms to avoid affecting the product in a negative manner; this poses a unique challenge when working with innovative biomaterials.
Brusco: Can you offer insight on Unique Device Identification (UDI) and its impact on packaging requirements?
Egan: The effects of UDI on packaging still has not filtered down to thermoformers in general, as most of the requirements come into effect after the trays and blisters have been delivered to the OEM.
Socola: We can only offer insight from the perspective of a validation lab. The reprocessing of an orthopedic device multiple times (i.e., clean and disinfect/sterilize) can affect the legibility and function of any labeling on the device itself. Therefore, OEMs should consider materials compatibility testing as part of design validation for any device that has the UDI directly on it to ensure that after multiple uses, this information is still legible and no adverse defects are noted. We have seen degradation to the UDI on devices in our lab after multiple exposures to the cleaning and sterilization process.
Whalen: The FDA has released a final ruling requiring that most medical devices distributed in the United States carry a unique device identifier, or UDI. It also applies to certain combination products that contain devices and those licensed under the Public Health Service. Compliance dates depend on classification of the device. Class III devices have the earliest requirement dates. A UDI system has the potential to improve the quality of information in medical device adverse event reports, which will help the FDA identify product problems more quickly, better target recalls, and improve patient safety. CleanCut is well positioned to support our customers in helping them meet these requirements with our various labeling and printing systems and equipment.
White: For OEMs, the amount of up-front work surrounding registration of devices, GTIN barcoding, overall label validation, and the potential impact on field inventory for rework and re-label as compliance deadlines approach can be cumbersome. Partnering with a packaging supplier to assist with the transition to serialized inventory will be an effective option for OEMs.
Brusco: What changes to orthopedic device packaging and sterilization do you see happening in the years to come?
Egan: Medical device manufacturers will continue to drive efficiencies in their processes to reduce costs. Packaging design must protect the device to point-of-use but needs to take into consideration “total cost of ownership” to the OEM through efficient design that eliminates material waste, reduces transport/storage costs and sterilization costs. Packaging will need to add functionality to the device and procedure by meeting the needs of the surgical team. For example, presenting all components required for the procedure in one handling tray or in aiding the clinician in prepping the device before introducing it to the patient.
Mach: There is a big push for recycling medical packaging right now. Although it’s in its early stages, in the future we will see large strides by manufacturers to designate materials as recyclable, and hospitals working toward recycling those materials—bringing about less impact on landfills.
Prust: Single-use device packaging will continue to become “greener” by reducing non-sustainable materials and dimensions. Combination devices are often humidity and temperature sensitive, which in some applications can be addressed by specific packaging design and component materials to help protect the product during transportation. Reusable device packaging will continue to migrate to containerized systems specific to the device with improved sealing and integrity detection systems.
Schwartz: In the future, we will continue to see greater need for faster, more cost-effective, and more nimble sterilization methods. For example, patient-specific implants manufactured with 3D printing need to be built and sterilized quickly and close to where the procedure will occur. Current sterilization methods rely on large batch manufacturing processes that don't fit that fast-turn model. Sterilization methods of the future will be scaled to both ends of the throughput volume requirement spectrum, enable in-line processing for quick turns and greater manufacturing efficiency, can be performed with parametric release, and offer a fast feasibility-to-validation process. We will also see the possibility for alternative packaging materials to Tyvek, as well as the ability to incorporate novel component materials, such as with combination devices.
Socola: For reusable orthopedic devices/sets processed within healthcare facilities, the packaging has evolved in recent times from wrapping the tray/set in two layers of a sterilization wrapper before sterilization, to placing the tray or set within a rigid sterilization container. Orthopedic manufacturers may recommend this practice for a number of reasons. One reason stated by the OEM is the weight of the tray or set itself, as many orthopedic trays/sets are fairly heavy and may cause some sterilization wraps to be breached or compromised by either abrasion (placed on and off shelves) or through compression (as when stacking sets on top of one another in sterile storage). Under these situations, the use of rigid sterilization containers may aid in the stacking and removal of trays and sets from storage shelves while helping to maintain package integrity. However, there have been significant changes to sterilization wraps in recent years as well, and this trend is likely to continue. There are now more companies selling SMS sterilization wrap in the North American healthcare market than ever before.
Whalen: More hospitals are now requesting that orthopedic medical device makers have their implants arrive individually sterilized. In the past, hospitals would autoclave entire trays of non-sterile implants prior to surgery. This practice, however, raises questions about the integrity of the implant following repeated sterilization cycles, a lack of traceability of implant batch, and most significantly, the correlation to post-operative infection and its costly consequences.
White: There will be an increase in the utilization of a single package configuration across multiple product designs. This provides a type of “pre-validated packaging” to increase time to market while reducing upfront costs. Single package designs that fit a vast majority of devices will help reduce the environmental footprint of packaging. Additionally, improved packaging equipment and methods will be vital for OEMs to improve costs and associated lead times.
Brusco: Is there anything else regarding orthopedic device packaging and/or sterilization you'd like to say?
Schwartz: Universal sterilization requirements and standards across the world would drive simpler regulatory approvals. This would be a vast improvement over each country having their own set of requirements.
Socola: OEMs can help themselves tremendously by putting together validation teams during product development and before device testing. I can’t tell you the number of times we received an orthopedic set for validation testing to only find out that the silicon tabs which tightly hold a device in place within the sterilization set are actually impeding sterilant penetration. The number of access holes in the tray itself and vent to volumes ratios also can have a dramatic effect of device sterilization. If these design issues are not addressed early enough, they can lead to sterilization efficacy failures and ultimately a redesign of the tray or set itself in one way or another. It most cases, design engineers working with experts in sterilization can make design changes early on in the process to significantly aid in getting the desired results during sterilization efficacy validation studies.
Whalen: Almost as important as the OEM medical or orthopedic device is its packaging. Today’s packaging is designed to be as unique and effective as the device itself. Much needs to be considered in the process. The packaging solution must maintain the sterile barrier system, protect the device in transit and use, and successfully communicate the OEM’s branding message. When this process is considered early, costly mistakes can be avoided and the ideal packaging solution can be created.
White: Allocating sufficient time for packaging and sterilization validations is essential to get products to market effectively and efficiently. Proper execution provides assurance of product quality and patient safety.
|
Niki Pidopiastis, Kevin O’Hara • SteriPro Consultants, Sterigenics John Kowalski • Independent Consultant, Sterigenics SteriPro Early orthopedic devices like plates and screws were almost exclusively metallic and made from high grade stainless steel, and readily sterilized by moist heat, ethylene oxide (EtO) gas, and ionizing radiation. In developing sterilization processes for metallic orthopedic devices, product developers were not concerned about damage to the device by the sterilization process due to the robust nature of the stainless-steel material. More concern was directed to the effect of the sterilization process on the packaging used for these devices. Many orthopedic devices currently on the market and under development are profoundly different with respect to both materials of construction and complexity of design. In addition to metals such as alloys of titanium, orthopedic implants now contain many types of polymers and engineering plastics, composites, absorbable polymers, biological materials, and embedded electronics. Many orthopedic device manufacturers do not have in-house sterilization capabilities and rely on one or more contract sterilization facilities. Such contract sterilizers process millions of cubic feet of medical products annually. For processing of large volumes of product, sterilization with EtO gas and ionizing radiation (mostly gamma irradiation) predominate. Because of the high temperature of moist heat sterilization, it is seldom used for orthopedic products with nonmetallic construction materials. Other processes are available for orthopedic device sterilization based on hydrogen peroxide, peracetic acid, nitrogen dioxide, chlorine dioxide, and seeded gas plasma. These processes might be ideal for low volume/high value products incompatible with EtO or ionizing radiation. A key aspect of a firm’s product development process (PDP) is the notion of “design for sterilization.” This should occur as early as possible in the PDP to help ensure straightforward, rapid, and successful validation of a preferred sterilization process. Many product development specialists and sterilization scientists believe exposure to ionizing radiation is the ideal sterilization method. It is cost competitive and there is a lot of volumetric processing capacity on a worldwide basis. It is conducted at relatively low temperature and does not involve exposure of the orthopedic device to toxic substances, residuals of which can remain on or in the product. Unacceptable changes/damage to certain device materials can occur; use of such materials can be avoided by designing for ionizing radiation sterilization in the PDP. Radiation-induced damage to materials can also be mitigated. EtO sterilization is generally mild with respect to materials effects and there is also a lot of volumetric capacity worldwide. Being highly toxic and a possible human carcinogen, effective aeration for residual removal is critical. Lower temperature processes have been developed to minimize damage to orthopedic and other devices coated with or containing a drug, biologic, or absorbable polymer, for example. For the other sterilization modalities mentioned, process temperature, sterilant residuals, and potential materials effects must be considered and evaluated. As with EtO and ionizing radiation, assessment of unacceptable changes to the product and/or package must be performed after processing and aging to show acceptability at the stated expiry date. In some cases, the ability to use a given sterilization process will be driven by its effect on a single product component, often a unique material, absorbable polymer, drug, or biologic. The PDP and sterilization process validation activities must always ensure each orthopedic implant comes out of sterilization with low probability of nonsterility (generally a SAL of 10-6 or better) and it and its packaging meet predetermined quality requirements throughout its shelf life. Sterilization processes have evolved and new ones have been developed to meet the challenges many new orthopedic devices pose. For ionizing radiation, several strategies have been introduced:
Manufacturers of orthopedic devices, particularly for devices with complex designs and sensitive materials/drugs/biologics, should consider sterilization as early as possible. If the device’s structure is not compatible with ionizing radiation, the biologic coating is not compatible with EtO sterilization, and neither is compatible with moist heat, this must be identified early to avoid “late bad news” (missed market release date, extra development/testing costs, etc.). Although sterilization processes have evolved and new ones have and will be introduced, product design and materials issues will, in most cases, determine the sterilization modality ultimately validated. |